
федеральное государственное автономное образовательное учреждение высшего образования Первый Московский государственный медицинский университет имени И.М. Сеченова Министерства здравоохранения Российской Федерации (Сеченовский Университет)

В.А. Парфенов, Н.Н. Яхно, Г.Ю. Евзиков
НЕРВНЫЕ БОЛЕЗНИ
Нервные болезни: Учебник/ В.А. Парфенов, Н.Н. Яхно, Г.Ю. Евзиков. — Москва : ООО «Издательство «Медицинское информационное агентство», 2018 г. |
Учебник
Рекомендовано Координационным советом по области образования «Здравоохранение и медицинские науки» в качестве учебника для студентов образовательных учреждений, реализующих программы высшего образования по специальности 31.05.03 «Стоматология» по дисциплине «Неврология»

Медицинское информационное агентство
Москва
2018
УДК 616.8(075.8)
ББК 56.1я73
П18
Парфенов, В.А. |
||
П18 |
Нервные болезни: Учебник/ В.А. Парфенов, Н.Н. Яхно, Г.Ю. Евзиков. — Москва : ООО «Издательство «Медицинское информационное агентство», 2 018 .-496 с.» |
|
ISBN 978-5-9986-0314-3 |
||
Учебник написан коллективом кафедры нервных болезней и нейрохирургии Первого МГМУ имени И.М. Сеченова. Описываются основные методы неврологического обследования, симптомы, синдромы и топическая диагностика поражений нервной системы. Излагаются этиология, патогенез, клиническая картина, диагноз, дифференциальный диагноз, консервативное и хирургическое лечение основных заболеваний нервной системы. Учебник направлен на формирование клинического мышления, способности поставить диагноз основных неврологических заболеваний, назначить эффективное лечение и провести своевременную профилактику. Клинические задачи и задания в тестовой форме позволят студентам и молодым врачам проверить усвоение учебного материала, подготовиться к зачету и экзамену. Содержание учебника соответствует требованиям Федерального государственного образовательного стандарта высшего образования по направлению подготовки 31.05.03 «Стоматология» и рабочей программе по дисциплине «Неврология». Для студентов стоматологического факультета медицинских вузов, клинических ординаторов и врачей-неврологов. |
||
УДК 616.8(075.8) ББК 56.1я73 |
ISBN 978-5-9986-0314-3 |
© Парфенов В.А., Яхно Н.Н., Евзиков Г.Ю., 2018 © Оформление. ООО «Издательство «Медицинское информационное агентство», 2018 © ФГАОУ ВО Первый МГМУ имени И.М. Сеченова Минздрава России (Сеченовский Университет), 2018 Все права защищены. Никакая часть данной книги не может быть воспроизведена в какой-либо форме без письменного разрешения владельцев авторских прав. |
20.2. Полиневропатии
Полиневропатии представляют группу заболеваний с одновременным множественным диффузным поражением периферических нервов. Они проявляются симметричными вялыми (периферическими) парезами (часто с преобладанием в дистальных отделах конечностей) и/или чувствительными нарушениями по полиневропатическому типу, вегетативно-трофическими расстройствами, иногда сочетаются с поражением черепных нервов.
Этиология. Основные причины полиневропатий представлены в табл. 20-1. Чаще всего (примерно в трети случаев всех полиневропатий) встречается диабетическая полиневропатия. У большинства людей, которые хронически злоупотребляют алкоголем, развивается алкогольная невропатия.
Метаболические нарушения |
Сахарный диабет, уремия, печеночная недостаточность, гипотиреоз, цирроз печени, подагра |
Алиментарные нарушения |
Недостаточное или несбалансированное питание (гиповитаминоз витаминов группы В), нарушение всасывания витамина B12 |
Токсические поражения |
Алкоголь (этиловый спирт), мышьяк, свинец, таллий, фосфорорганические соединения, осложнения после приема лекарственных средств (изониазид, дифенин, талидомид, нитрофураны) |
Инфекционные заболевания |
Дифтерия, лепра, ВИЧ-инфекция, боррелиоз, ботулизм, паротит, тиф и паратиф |
Наследственные полиневропатии |
Моторно-сенсорные или сенсорно-моторные полиневропатии, невропатии со склонностью к параличам от сдавления, полиневропатия при острой печеночной порфирии, полиневропатия при первичном (семейном) амилоидозе |
Другие причины |
Злокачественные новообразования (паранеопластические синдромы), диспротеинемии (парапротеинемии), заболевания соединительной ткани (узелковый периартериит, ревматоидный артрит, системная красная волчанка, склеродермия), саркоидоз |
Клиническая картина. Полиневропатия обычно возникает постепенно. Начальные признаки — парестезии, снижение тактильной и болевой чувствительности в пальцах стоп и/или кистей, возможны невропатические боли. В дальнейшем зона гипестезии поверхностной чувствительности приобретает форму носков и/или перчаток, снижается глубокая чувствительность, ослабевают или полностью утрачиваются сухожильные, особенно ахилловы, рефлексы, развиваются вялые атрофические парезы преимущественно в дистальных отделах конечностей. Чаще симптомы преобладают в ногах.
Диагноз полиневропатии основывается на клинических и инструментальных данных. Поражение периферических нервов подтверждают результаты электронейромиографии: симметричное замедление скорости проведения возбуждения по периферическим нервам, снижение амплитуды мышечного ответа. Вдиагностически неясных ситуациях проводят морфологическое исследование (биопсию) мышцы (определение нейрогенного или мышечного поражения) или нерва, что позволяет уточнить характер и причину его поражения.
Выявив полиневропатию, необходимо установить ее причину, что требует тщательного соматического обследования больного.
Лечение полиневропатии зависит от ее этиологии. При экзогенных причинах (интоксикация, гиповитаминоз) необходимо их устранение. В случаях соматического заболевания ведущее значение приобретает его лечение. При двигательных нарушениях рекомендуют лечебную гимнастику, массаж, физиотерапевтические процедуры, при грубом парезе стоп — ношение ортопедической обуви.
20.2.1. Острая воспалительная демиелинизирующая полирадикулоневропатия (синдром Гийена-Барре)
Развитие заболевания связывают с аутоиммунным повреждением миелина периферической нервной системы, приводящим к сегментарной демиелинизации в передних корешках, проксимальных отделах периферических нервов, сплетениях. Распространенность заболевания составляет 1—4 случая на 100 тыс. населения.
Клиническая картина. У половины больных за 1—3 недели до появления неврологических симптомов отмечаются инфекционные заболевания верхних дыхательных путей или желудочно-кишечного тракта. Первые неврологические нарушения — боли и парестезии в ногах и/или парезы в них. В дальнейшем наблюдается прогрессирование чувствительных и двигательных расстройств в ногах и присоединение нарушений в руках. Симптомы нарастают в течение 1-4 недель. Ведущий признак заболевания — вялые параличи, которые часто распространяются в восходящем направлении, сначала захватывая мышцы ног и тазового пояса, затем туловища, шеи и иногда дыхательную мускулатуру, мышцы лица (восходящий паралич). Обычно первой поражается проксимальная группа мышц ног, затем мышцы туловища, межреберные мышцы, мышцы рук, шеи и головы, прежде всего лица.
В части случаев развиваются опасные для жизни дыхательные нарушения (вследствие паралича диафрагмы и межреберных мышц) и расстройство глотания. Черепные нервы поражаются в 75%, в половине из них страдает лицевой нерв.
В качестве осложнений, обусловленных в значительной степени обездвиженностью больного, возможны пролежни, пневмонии, тромбоэмболия легочной артерии, тромбозы вен. Летальность в настоящее время (при правильной врачебной тактике) не превышает 5%. У большинства больных спонтанное восстановление начинается спустя 10—12 дней после прекращения прогрессирования заболевания и продолжается в течение от 6 месяцев до 3лет. Полное восстановление наблюдается в половине случаев, у большинства остальных больных сохраняются только легкие неврологические расстройства илишь у 10—15% остаются грубые парезы, приводящие к стойкой инвалидности.
Редко (только в 3%) заболевание повторяется один или несколько раз.
Диагноз основывается на клинических данных: быстрое развитие периферических парезов конечностей, туловища и лица. При исследовании цереброспинальнойжидкости выявляют повышение содержания белка при незначительном увеличении клеток (белково-клеточная диссоциация). Электронейромиография обнаруживает снижение скорости и блоки проведения возбуждения по периферическим нервам в 90%, что указывает на демиелинизацию.
Лечение направлено главным образом на поддержание дыхания и сердечной деятельности при их нарушении, профилактику возможных осложнений ивоздействие на аутоиммунный процесс — плазмаферез, проведенный в первые 7 дней заболевания; рекомендуется 3—5 сеансов плазмафереза через день с обменом 1,5—2 л плазмы за сеанс. Эффективно в/в медленное (в течение 6—8 часов) введение иммуноглобулина из расчета 0,4 г/кг в 1 л физиологического раствора в течение 5 дней. Крайне важен общий уход за больным, оценка жизненной емкости легких и их искусственная вентиляция при дыхательной недостаточности, парентеральное (зондовое) питание при нарушении глотания. Искусственная вентиляция легких показана при первых симптомах дыхательной недостаточности или ателектаза (парциальное давление кислорода менее 70 мм рт. ст.) или при значительном снижении жизненной емкости легких (менее 12—15 мл/кг).
20.2.2. Наследственные полиневропатии
Заболевания носят семейный характер, но нередко встречаются спорадические его случаи. Большинство наследственных полиневропатий дебютирует в детском, юношеском или молодом возрасте, тем не менее часть из них — в зрелом и даже пожилом возрасте. Для установления диагноза могут потребоваться тщательный сбор семейного анамнеза, обследование родственников.
Моторно-сенсорные полиневропатии встречаются значительно чаще, чем сенсорные.
Возможны аутосомно-доминантный, аутосомно-рецессивный и сцепленный с Х-хромосомой тип наследования моторно-сенсорной полиневропатии.
Среди наследственных моторно-сенсорных полиневропатий наиболее часто встречается болезнь Шарко—Мари—Тута, которая ранее называлась невральной (перонеальной) амиотро- фией, или наследственной моторно-сенсорной полиневропатией I и II типов. Частота заболевания достигает трех случаев на 100 тыс. населения, она имеет несколько вариантов генетических дефектов, типов наследования и клинического течения.
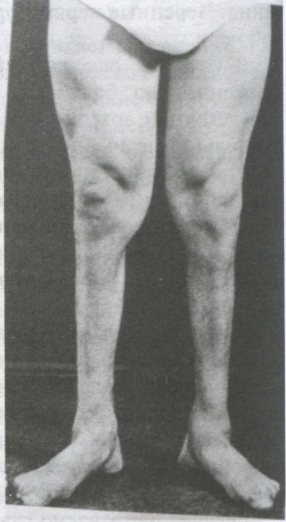
Клиническая картина. Симптомы болезни Шарко—Мари—Тута 1Атипа (мутация гена РМР22 на коротком плече 17-й хромосомы) обычно возникают в возрасте до 30 лет (чаще на первом или втором десятилетии) в виде медленно нарастающей симметричной слабости в дистальных отделах ног с развитием выраженных амиотрофий, через 5—10 лет присоединяется слабость и в дистальных отделах рук. Мышечная гипотрофия редко распространяется выше нижней трети бедер и локтевых суставов, внешне ноги могут напоминать «перевернутые бутылки от шампанского» (рис. 20-4).
Другие типы моторно-сенсорных полиневропатий отличаются по генетическому дефекту, времени дебюта, клиническим проявлениям, течению и прогнозу. При некоторых из них, помимо полиневропатии, отмечаются и другие неврологические нарушения (мозжечковая атаксия, атрофия зрительного нерва).
Диагноз конкретного типа наследственной полиневропатии устанавливается на основании клинико-инструментального обследования, генетического тестирования.
Лечение симптоматическое. Лечебная гимнастика, ортопедические вмешательства (ношение ортопедической обуви, хирургические вмешательства), правильная профессиональная ориентация способствуют поддержанию самостоятельного передвижения и самообслуживания, социальной и бытовой адаптации.