Медицинская генетика : национальное руководство / под ред. Е. К. Гинтера, В. П. Пузырева, С. И. Куцева. - Москва : ГЭОТАР-Медиа, 2022. - 896 с. (Серия "Национальные руководства") - ISBN 978-5-9704-6307-9 |
Аннотация
Настоящее издание подготовлено коллективом авторов, абсолютное большинство которых хорошо известны не только отечественному, но и зарубежному читателю.
В книге представлены основные разделы генетики человека и медицинской генетики, такие как генетика развития человека, эпигенетика, генетика соматических клеток, популяционная генетика, фармакогенетика, иммуногенетика и система генов HLA. Также описаны современные представления об основных классах наследственных болезней: моногенных, включая популяционную генетику моногенных заболеваний, наследственных болезнях обмена веществ, митохондриальных заболеваниях, патологиях, обусловленных расширением зоны тандемных тринуклеотидных повторов, хромосомных болезнях, включая те, что обусловлены микрохромосомными перестройками, заболеваниях, связанных с геномным импринтингом, многофакторных болезнях, врожденных пороках развития и тератогенных синдромах.
Отдельная часть руководства посвящена лечению и профилактике наследственных болезней, а также этическим проблемам медицинской генетики. В ней подробно рассмотрена проблема генной терапии, в том числе с использованием методов редактирования генома, которые многими исследователями рассматриваются как наиболее перспективные в генотерапии заболеваний человека.
Книга будет полезна врачам практически всех специальностей с учетом все возрастающего вклада генетики в современную медицину, без которого невозможно ее дальнейшее совершенствование.
Глава 15. Молекулярно-генетические методы диагностики наследственных болезней
Введение
Некоторая часть генетического груза вида Homo sapiens sapiens связана с заболеваниями, возникновение и развитие которых являются следствием изменения(й) в наследственном аппарате клеток. Более 70% патологий человека представлено мультифакторными, или полиэтиологическими, болезнями - это болезни, которые обусловлены как влиянием окружающей среды, так и изменениями генетического материала. Особенностями данной группы болезней являются высокая популяционная частота, выраженный клинический полиморфизм, неменделевское наследование, зависимость проявлений болезни от факторов внешней среды, возраста и пола больного. С генетической точки зрения характерной чертой данной группы заболеваний является то, что генетическим фактором риска является носительство вариантов нескольких (многих) генов одновременно, что делает практическое определение вероятности развития данной патологии для конкретного индивида весьма неточным. К наиболее частым мультифакторным болезням относят сахарный диабет, бронхиальную астму, ИБС, псориаз, шизофрению и многие другие.
До 10% наследственных патологий обусловлено изменениями числа или структуры хромосом. В настоящее время известно более 5 000 различных заболеваний, связанных с геномными и хромосомными мутациями.
18% приходится на моногенные болезни. Причиной данной группы заболеваний являются изменения ДНК на уровне генов. Необходимым и достаточным условием возникновения клинического фенотипа моногенной болезни является наличие мутаций в причинном гене. Однако клинические проявления болезни могут зависеть от пола, возраста больного, а манифестация заболевания - быть связана с неблагоприятными условиями внешней среды - травмой, переохлаждением, интенсивными физическими нагрузками. Данные болезни наследуются в соответствии с законами Менделя и также называются менделирующими болезнями. На середину 2021 г. в базе данных OMIM (Online Mendelian Inheritance in Man) описано 5872 клинических фенотипов, для 4106 из них известен молекулярный базис. Для большинства моногенных болезней характерна генетическая гетерогенность - за развитие одного и того же клинического фенотипа в неродственных семьях могут отвечать мутации различных генов. Среди моногенных болезней выделяют частые, встречающиеся с частотой более чем 1/10 000, - ФКУ, муковисцидоз, адреногенитальный синдром, и редкие - частота менее 1/100 000. Каждый человек является носителем в среднем пяти рецессивных мутаций, не проявляющихся при наличии второй нормальной копии гена. Именно моногенные болезни являются основным объектом ДНК-диагностики.
ДНК-диагностика - это поиск генетической причины болезни на уровне изменений ДНК. ДНК-диагностика направлена на поиск непосредственной причины наследственной болезни - патогенных вариантов гена и является наиболее адекватным и точным исследованием при моногенной патологии. Проведение данной диагностики возможно, даже если неизвестен точный патогенез развития болезни, но известен ген, который ее вызывает.
В зависимости от поставленных задач различают следующие виды ДНК-диагностики:
-
подтверждающая - проводится исследование ДНК больного человека с целью определения непосредственной причины болезни;
-
пресимптоматическая - исследуется ДНК клинически здоровых родственников больного, как правило, при наследственных болезнях с поздней манифестацией, таких как хорея Гентингтона;
-
диагностика носительства - проводится здоровым родственникам больных с рецессивными заболеваниями с целью планирования деторождения;
-
ПД - поиск мутации, ответственной за болезнь в семье, в материале плода - ворсинах хориона, амниотической жидкости, пуповинной крови на различных сроках беременности. Позволяет определить статус плода и решить вопрос о пролонгировании беременности на ранних сроках гестации 10-12 нед;
-
преимплантационная диагностика - исследование клеток эмбриона в рамках процедуры ЭКО с целью отбора зародышей, не несущих мутантный ген;
-
неинвазивные пренатальные тесты (НИПТ) - поиск мутаций в генетическом материале плода, циркулирующем в кровотоке матери.
В зависимости от объекта исследования выделяют косвенную и прямую ДНК-диагностику. При косвенной ДНК-диагностике исследуют не непосредственную причину заболевания, а определяют наследование в семье участка хромосомы, несущей предположительно мутантный ген по генетическим маркерам, лежащим в непосредственной близости от этого гена (рис. 15-1, см. цв. вклейку). Точность данного типа ДНК-диагностики составляет 95-99%, однако для его применения необходимы уверенность в клиническом диагнозе и знание того, что за данный клинический фенотип могут быть ответственны мутации только одного гена. Так как практически для всех заболеваний описана генетическая гетерогенность, использование косвенной диагностики как единственного метода нецелесообразно. На сегодняшний день возможно ее применение в семьях с точно установленной генетической причиной болезни для повышения точности пренатальной или преимплантационной генетической диагностики.
Прямая ДНК-диагностика направлена на поиск мутации, являющейся непосредственной причиной болезни. Точность такой диагностики составляет 100%. Возможно проведение прямой ДНК-диагностики при сомнительном диагнозе и при наличии нескольких локусов заболевания. Прямая ДНК-диагностика позволяет диагностировать носительство мутации в семьях с моногенной патологией даже при отсутствии материала больного.
Методы молекулярно-генетической диагностики
Материалом для проведения диагностики являются нуклеиновые кислоты, выделенные из ядерных клеток. Наиболее удобным материалом для исследования является цельная кровь в пробирке с антикоагулянтом или высушенная на фильтровальной бумаге. В связи с тем, что некоторые химические элементы ингибируют работу фермента ДНК-полимеразы, для проведения ДНК-исследования подходят только антикоагулянты этилендиаминтетраацетат (ЭДТА) или цитрат натрия. Кроме того, ингибировать ПЦР может большое количество чужеродной, например, бактериальной ДНК. Выделение ДНК производят различными методами в зависимости от типа предоставленного материала и метода молекулярно-генетического анализа, планируемого для исследования.
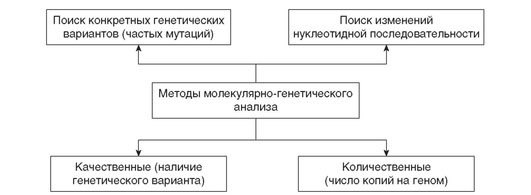
Можно выделить две группы методов ДНК-диагностики: методы, позволяющие исследовать только конкретные уже известные мутации, и методы, позволяющие найти любое изменение нуклеотидной последовательности ответственного за болезнь гена, в том числе и ранее не описанную мутацию (методы чтения нуклеотидной последовательности). По типам детектируемых вариантов можно выделить качественные (выявление наличия/отсутствия генетического варианта у обследуемого) и количественные методы (определение количества копий исследуемого фрагмента в геноме) (рис. 15-2).
Большинство методов, применяемых на сегодняшний день, использует свойство фермента ДНК-полимеразы обеспечивать многократное избирательное копирование целевого участка ДНК, ограниченного искусственно синтезированными последовательностями нуклеотидных затравок (праймеров), - ПЦР.
Наиболее распространенные методы, применяемые для поиска известных мутаций
Методы исследования уже известных мутаций возможно применять для болезней, гены которых хорошо изучены, определены частоты и спектры мутаций, выявлены мажорные мутации. Применение систем детекции частых мутаций позволяет удешевить и оптимизировать процедуру проведения ДНК-диагностики за счет снижения материально-технических и временных затрат. Системы детекции наиболее частых мутаций успешно применяются в молекулярно-генетической диагностике частых аутосомно-рецессивных заболеваний: СМА, муковисцидоза, ФКУ, несиндромальной нейросенсорной тугоухости, адреногенитального синдрома и других. На рис. 15-3 показана система детекции в одной пробирке восьми наиболее частых мутаций гена РАЯ, ответственного за ФКУ.
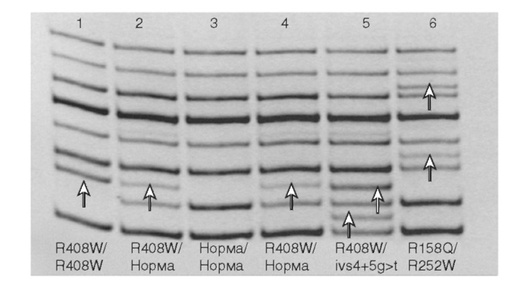
Такая же система детекции мутаций может быть использована для выявления мутаций при ряде аутосомно-доминантных болезней, при которых показано наличие мажорной мутации. Например, причиной 99% случаев ахондроплазии является мутация Gly380Arg гена FGFR3, которая возникает в результате замены 1138 нуклеотида гуанина на аденин или цитозин.
Принцип метода: исследование разницы длин целевого фрагмента после амплификации с использованием разделения по длине с помощью электрофореза в различных типах гелей.
Область применения.
-
Поиск небольших делеций/инсерций, поиск экспансий, исследование STR-маркеров с целью определения родства или изучения сегрегации определенного локуса в семьях, КФ-ПЦР для диагностики анеуплоидий.
-
Метод исследования полиморфизма длин рестрикционных фрагментов применим для исследования известных генетических вариантов и является качественным.
Принцип метода основан на свойстве эндонуклеаз рестрикции (рестриктаз) «узнавать» строго определенную нуклеотидную последовательность (сайт рестрикции) и разрезать молекулы ДНК в местах узнавания. Фермент подбирается таким образом, чтобы исследуемый вариант нуклеотидной последовательности изменял сайт узнавания рестриктазы. После обработки продукта ПЦР эндонуклеазой проводится исследование разницы длин полученных фрагментов.
Область применения. Поиск известных вариантов нуклеотидной последовательности.
Принцип метода: основан на свойстве фермента Pfu-лигазы высокоспецифично сшивать нуклеотидные разрывы комплементарных матрице последовательностей. Для детекции точковых мутаций создаются нуклеотидные конструкции (пробы), комплементарные норме или варианту нуклеотидной последовательности. При этом в каждую пробу вводят различную по длине маркерную последовательность, чтобы различать отдельные аллели по длине, а концы проб замыкают универсальными праймерами. В результате амплификации лигированных проб получают набор продуктов с длинами, соответствующими различным аллелям. На рис. 15-4 (см. цв. вклейку) представлена принципиальная схема метода. Аллельспецифичная лигаза-зависимая амплификация проб является быстрым высокоспецифичным методом. Основным преимуществом данной методики детекции вариантов нуклеотидной последовательности является возможность мультиплексирования, т.е. детекции в одной пробирке 10 вариантов нуклеотидной последовательности и более, независимо от их положения в геноме друг относительно друга. Использование в системе флюоресцентно меченного праймера позволяет детектировать результаты реакции с использованием капиллярного электрофореза и сочетать, при необходимости, в одной пробирке детекцию точковых вариантов и количественный анализ.
Область применения. Поиск известных точковых замен нуклеотидной последовательности.
Методы количественного анализа
Все методы ДНК-диагностики можно разделить на качественные и количественные. Качественные методы способны выявить точковое изменение нуклеотидной последовательности, короткие делеции и инсерции, протяженные делеции генов, имеющих в геноме одну копию (гены, локализующиеся на половых хромосомах мужчин), - к таким методам относятся анализ полиморфизма длин рестрикционных фрагментов, анализ полиморфизма длин амплификационных фрагментов, прямое автоматическое секвенирование и другие. Количественные методы позволяют определить число копий гена в геноме. К таким методам относятся блотгибридизация, ПЦР в реальном времени, количественная мультиплексная лигазная реакция. Использование данных методов необходимо при поиске протяженных делеций и дупликаций генов, лежащих на аутосомах, например, детекции дупликаций/делеций гена PMP22, ответственного за наследственную моторно-сенсорную нейропатию 1А типа, но особенно важно при исследовании носительства генов заболеваний, мажорными мутациями которых являются делеции, например спинальной мышечной амиотрофии или мышечной дистрофии Дюшенна-Беккера.
Принцип метода основан на детекции продукта ПЦР во время экспоненциальной фазы ПЦР в режиме реального времени с использованием специфичных флюоресцентных зондов или интеркалирующих красителей, которые способны флюоресцировать при наработке продукта амплификации. Метод позволяет произвести количественную оценку содержания искомой матрицы в образце. Использование зондов с различными флюорофорами позволяет анализировать несколько вариантов последовательности ДНК в одной пробирке.
Область применения: поиск известных точковых вариантов нуклеотидной последовательности, анализ протяженных делеций, инсерций, исследование экспрессии генов.
Принцип метода: количественное определение продукта ПЦР со сшитых лигазой специфически-гибридизованных проб. Так как фермент лигаза обладает высокой специфичностью, метод позволяет достоверно различать генотипы «2:2» и «2:3», «2:3» и «2:4», например, при определении числа копий генов локуса SMN. Метод позволяет анализировать для одного тестируемого образца ДНК до 50 проб, комплементарных разным участкам генома в одной пробирке.
Область применения: поиск протяженных делеций и дупликаций в геми, гомо- и гетерозиготном состоянии, анализ анеуплоидий, определение числа копий генов и псевдогенов на геном.
Принцип метода заключается в полногеномной амплификации с последующей гибридизацией на чипах-матрицах, содержащих различное количество целевых точек, относительно равномерно распределенных по геному. Разрешающая способность метода зависит от плотности распределения точек и их числа. Позволяет анализировать CNVs в ДНК тестируемого образца по сравнению с эталонным образцом.
Область применения: выявление несбалансированных хромосомных аномалий.
Выявление микроделеций/микродупликаций, размер которых превышает разрешающую способность чипа (обычно 50 000-100 000 п.н., для ХМА экзонного уровня разрешающая способность составляет 2-3 экзона гена на уровне последовательности ДНК).
Методы, позволяющие исследовать всю последовательность гена
Для поиска причин редких болезней и болезней, в генах которых не выявлено мажорных мутаций, применяются методы, позволяющие исследовать всю кодирующую последовательность и регуляторные области соответствующего гена, некоторый набор генов (панель) или весь геном.
На сегодняшний день является референсным методом определения нуклеотидной последовательности фрагмента ДНК.
Принцип метода. Исследуемый фрагмент нуклеотидной последовательности выступает матрицей для синтеза новой цепи с использованием фермента ДНК-полимеразы. Кроме обычных дезоксинуклеотидтрифосфатов, в реакционную смесь добавляют «терминаторы» - флюоресцентно-меченные дидезоксину-клеотидтрифосфаты, случайным образом обрывающие синтез цепи при присоединении к комплементарному нуклеотиду. Полученное в результате сиквенсовой реакции множество фрагментов детектируется с использованием капиллярного электрофореза (рис. 15-5, см. цв. вклейку)
Область применения: поиск вариантов нуклеотидной последовательности в фрагментах конкретных генов, референсный метод для валидации изменений, выявленных массовым параллельным секвенированием.
Генетическая гетерогенность наследственных болезней и большой размер некоторых генов приводят к тому, что секвенирование всех ответственных за данный клинический фенотип нуклеотидных последовательностей является дорогостоящей и длительной процедурой. На сегодняшний день для таких заболеваний появилась возможность применения в диагностической практике технологий массового параллельного секвенирования (секвенирования следующего поколения, высокопроизводительного секвенирования, NGS), позволяющих одномоментно получить массив данных, содержащих информацию о нуклеотидной последовательности нескольких целевых генов или всего экзома и генома.
Методы массового параллельного секвенирования можно разделить на две большие группы: методы с предварительной клональной амплификацией фрагментов ДНК и методы секвенирования одиночных молекул ДНК.
Секвенирование с использованием клональной амплификации библиотек на сегодняшний день широко используются. Среди технологий, предполагающих амплификацию фрагментов ДНК, можно выделить следующие.
-
Секвенирование лигированием предварительно иммобилизованных на твердой фазе клональных библиотек коротких одноцепочечных исследуемых фрагментов с флюоресцентно-меченными короткими вырожденными нуклеотидными последовательностями, таким образом, что каждому флюорофору соответствует определенный нуклеотид в определенной позиции. Проводят 10-15 последовательных лигирований и производят «перезагрузку» с присоединением следующего праймера со сдвигом на одну букву.
-
Технология секвенирования синтезом с использованием обратимо-терминирующих нуклеотидов, основана на регистрации сигнала, образующегося при формировании фосфодиэфирной связи в реакции присоединения определенного нуклеотида (аденина, тимина, гуанина или цитозина) ферментом полимеразой, сигнал считывается после присоединения обратимо-терминированного на 3'-конце флюоресцентно-меченного динуклеотидтрифосфата. После детекции флюорофор и блокатор удаляют из синтезируемой цепи и присоединяют следующий нуклеотид.
-
Полупроводниковое секвенирование по принципу напоминает пиросеквенирование, однако присоединение определенного нуклеотида регистрируется по образующимся в результате этого присоединения ионам водорода. Преимуществом данного метода является отсутствие оптического детектора сигнала, что существенно упрощает и удешевляет прибор.
Секвенирование одиночных молекул ДНК является перспективным подходом, позволяющим получить длинные прочтения до 100 000 п.н., что необходимо при сборке ранее неизвестных геномов или прочтении повторяющихся областей. На сегодняшний день развиваются следующие технологии: секвенирование синтезом с использованием обратимо-терминирующих нуклеотидов без этапа создания клональной библиотеки; регистрация изменений ионного тока, вызванных прохождением молекул ДНК через нанопоры под действием электрического тока; детекция спектра комбинированного рассеяния света с использованием атомного микроскопа и другие. Несмотря на то что пока разработчикам не удалось добиться высокой точности при определении нуклеотидного состава одиночных молекул ДНК, секвенирование протяженных последовательностей востребовано и используется для сборки новых геномов, а в области медицинской генетики - для изучения областей повторов.
Последовательность массового параллельного секвенирования включает следующие этапы.
-
Фрагментация исходных образцов ДНК с получением фрагментов заданной длины.
-
Присоединение по краям фрагментов синтетических олигонуклеотидных адаптеров.
-
Создание библиотек случайных фрагментов - набора фрагментированных, модифицированных и амплифицированных фрагментов с помощью эмульсионной ПЦР (разделение колоний с использованием капель) или мостиковой ПЦР (разделение колоний на поверхности стекла).
-
Определение нуклеотидной последовательности библиотек фрагментов.
Особенность методов массового параллельного секвенирования заключается в огромном вкладе биоинформатических методов в процесс получения конечного результата молекулярно-генетического анализа. Результатом всех приведенных выше действий является несколько миллионов коротких прочтений, которые необходимо собрать в длинные последовательности и провести анализ. Приборы для секвенирования в качестве входящего сигнала получают оптический или электрический сигнал, соответствующий определенному нуклеотиду, который преобразуют для исследователя в последовательность нуклеотидов с приписанным каждому из нуклеотидов значению достоверности полученного результата. Данные значения прибор сохраняет в файл определенного типа, наиболее распространенным из которых является формат FASTQ.. Расчет вероятности ошибки в каждой из позиций проводят на основании данных о секвенировании известных последовательностей. Калибровку для каждого типа приборов проводит производитель по собственной технологии.
В области медицинской генетики нет необходимости собирать заново каждый прочтенный геном, достаточно картировать полученные прочтения на референсный геном человека. Хотя картирование является не столь простой задачей: необходимо учитывать наличие в прочтенной последовательности множества отличий, связанных с особенностями конкретного индивида и ошибками прочтений генома, разработано множество математических алгоритмов и программ для выравнивания успешно с ней справляющихся. Для того чтобы отфильтровать случайные ошибки детекции нуклеотидов, каждое место генома прочитывается многократно в обе стороны. После картирования и выявления вариаций генетической последовательности в образце исследуемого человека необходимо их правильно «назвать», т.е. привязать к конкретной геномной координате и оценить патогенность выявленных изменений. На первом этапе проводится автоматический поиск выявленных вариантов в различных генетических базах данных (аннотация): dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/), OMIM (https://www.ncbi.nlm.nih.gov/omim), gnomAD (https://gnomad.broadinstitute.org/), COSMIC (https://cancer.sanger.ac.uk/cosmic) и т.д. В результате аннотирования файлов исследователь получает файл в формате VCF, который содержит информацию обо всех выявленных отличиях исследуемого образца от референсной последовательности, геномные координаты, сведения о числе прочтений (ридов), на которых встретилось данное изменение, и т.д.
Большинство используемых на сегодняшний день сервисов для анализа данных массового параллельного секвенирования проводит оценку патогенности с использованием программ-предсказателей патогенности: SIFT (https://sift.bii.a-star.edu.sg/), PROVEAN (http://provean.jcvi.org/index.php) и др. и ранжируют выявленные варианты.
На завершающем этапе исследования оценку патогенности и возможной взаимосвязи с фенотипом больного выявленных в результате анализа и автоматической фильтрации вариантов проводит врач лабораторный генетик с использованием дополнительных баз данных, программ предсказания и клинической информации, предоставленной для проведения исследования. Все варианты нуклеотидной последовательности интерпретируются в соответствии с Руководством по интерпретации данных последовательностей ДНК человека, полученных методами массового параллельного секвенирования, редакция используемых рекомендаций указывается в заключении. Обобщением результата анализа является заключение по результатам массового параллельного секвенирования. Необходимо отметить, что количество информации о генетических причинах менделирующих заболеваний постоянно растет, поэтому данные NGS могут быть многократно проанализированы по мере накопления новых знаний.
К сожалению, полное секвенирование геномов остается достаточно дорогостоящим методом исследования, поэтому в практической медицинской генетике наиболее распространено исследование панелей генов различной величины.
Для того чтобы выделить и прочитать только целевые последовательности некоторого набора генов (панели), набора генов, при которых описаны фенотипы болезней (клинического экзома), или всех кодирующих последовательностей генома (экзома), существуют технологии целевого обогащения библиотек фрагментов ДНК. Наиболее распространенными являются подходы на основе мультиплексной ПЦР с праймерами на целевые фрагменты генома и подходы на основе гибридизации с последующей ПЦР. Предпочтительная технология обогащения выбирается в зависимости от поставленной задачи, размера таргетного региона, предполагаемого количества и качества исходной ДНК, сложности процесса пробоподготовки и так далее.
При выборе методов на основе массового параллельного секвенирования в качестве диагностических необходимо помнить об ограничениях.
Любое секвенирование панели генов не позволяет выявлять инсерции и делеции длиной более 20% длины прочтения (при длине прочтения 75 п.н. - это 15 пар оснований), мутации в интронных областях (за исключением канонических сайтов сплайсинга), вариации длины повторов (в том числе экспансии триплетов), а также мутации в генах, у которых в геноме существует близкий по последовательности паралог (псевдоген). Метод не предназначен для определения цис-, транс-положения пар гетерозиготных мутаций [однако цис/транс-положение может быть определено в случае близкого (на длине одного прочтения) расположения вариантов], выявления хромосомных перестроек, полиплоидии, выявления мутаций в состоянии мозаицизма. Кроме того, многие технологии массового параллельного секвенирования имеют погрешности при исследовании гомополимерных областей.
Используя секвенирование генома, возможно проводить анализ числа копий генов (CNV), сложных структурных перестроек, а в некоторых случаях - и анализ числа повторов.
Методы секвенирования одиночных последовательностей позволяют прочитать длинные повторяющиеся последовательности, но пока обладают высоким числом ошибок определения нуклеотидной последовательности.
Показания для назначения молекулярно-генетической диагностики
-
Наличие клинических проявлений менделирующего заболевания. В данном случае проведение диагностики необходимо для подтверждения диагноза на молекулярно-генетическом уровне и выработки тактики профилактики наследственной патологии в семье.
-
Выявление у пробанда при рутинных обследованиях биохимических, электрофизиологических и других изменений, характерных для моногенной патологии. Несмотря на то что некоторые диагнозы могут быть подтверждены на основании выявления типичных биохимических маркеров, например, специфичного повышения концентрации фенилаланина в сыворотке крови у больных ФКУ, проведение ДНК-диагностики выявит молекулярную причину заболевания, в сомнительных случаях даст ответ о верности диагноза, сделает возможным проведение в семье диагностики носительства и ПД. Существуют болезни, при которых выявляются характерные, но не специфичные биохимические изменения, например, повышение уровня креатинфосфокиназы и трансаминаз возможно при ряде ненаследственных заболеваний и моногенных заболеваниях: мышечных дистрофиях, кардиомиопатиях, изолированном повышении уровня креатинфосфокиназыв сыворотке крови и других. В таких случаях ДНК-диагностика - единственный способ окончательно подтвердить диагноз, установить молекулярную природу болезни, определить тип наследования в изолированных случаях, определить риски рождения больного ребенка и выработать тактику профилактики наследственной патологии в отягощенных семьях. В некоторых ситуациях подтверждающая диагностика с использованием молекулярно-генетических методов проще, быстрее и дешевле, чем проведение комплекса диагностических мероприятий при подозрении на болезнь, например, при стойком повышении непрямого билирубина для диагностики синдрома Жильбера сегодня оптимально использовать тест поиска инсерции в промоторной области гена UGT1, в отличие от функциональных тестов с индукторами уридинфосфатглюкуронозилтрансферазы I в условиях стационара. Повсеместное внедрение аудиологического скрининга новорожденных позволяет выявить нейро-сенсорную тугоухость в первые дни жизни ребенка. Для успешной реабилитации и определения рисков повторения в семье патологии необходимо установить природу нарушений слуха. Эффективным диагностическим тестом в данном случае является поиск мутаций в гене GJB2 - выявление двух мутаций позволяет вовремя провести кохлеарную имплантацию без дополнительных исследований.
-
Наличие подтвержденной моногенной патологии у кровных родственников пробанда.
-
Поиск носительства частых рецессивных заболеваний у пар, планирующих деторождение. Некоторые рецессивные болезни, например ФКУ, муковисцидоз, адреногенитальный синдром и др., имеют высокую частоту гетерозиготного носительства практически на всех континентах. Высокая частота носительства других рецессивных заболеваний характерна лишь для представителей определенных популяций, например, носительство периодической болезни у средиземноморских народов или рецессивного остеопетроза у чувашей. При планировании парой беременности можно рекомендовать обследоваться на носительство распространенных рецессивных болезней для выявления пар высокого риска и принятия своевременных профилактических мер.
-
Выявление признаков моногенной патологии плода при обследовании беременной неинвазивными методами. Сделать предположение о возможном наличии моногенной патологии у плода даже в случае отсутствия в родословной семьи наследственных заболеваний можно при проведении неинвазивных обследований. Например, при проведении УЗИ можно выявить маркеры, свидетельствующие об ахондроплазии или поликистозе почек плода. В данных случаях возможно проведение инвазивной пренатальной диагностики с целью поиска мутаций непосредственно у плода.
-
Проведение пренатальной или преимплантационной ДНК-диагностики при беременностях в семьях с высоким риском моногенной патологии.
После ДНК-диагностики лабораторией выдается заключение, в котором указывается ген или конкретные варианты одного или нескольких генов, которые были исследованы у пробанда, метод исследования и результат диагностики. В случае исследования методами массового параллельного секвенирования приводятся сведения о качестве исследования.
При проведении ДНК-диагностики могут быть выявлены следующие виды изменений нуклеотидной последовательности: патогенный вариант, вероятно патогенный вариант, вариант неопределенного клинического значения, вероятно доброкачественный вариант, доброкачественный вариант. Патогенность варианта определяется в соответствии с критериями патогенности и может быть пересмотрена при накоплении дополнительной информации: косегрегации варианта и заболевания в семье, статуса de novo, цис-, транс-положения вариантов при рецессивном заболевании, появлении результатов функционального анализа, данных о популяционных частотах варианта и встречаемости в выборках больных.
В заключение молекулярно-генетического анализа должны выноситься только патогенные, вероятно патогенные и варианты неопределенного клинического значения. Согласно рекомендациям по интерпретации данных геномных исследований, ПД можно проводить только в отношении патогенных и вероятно патогенных вариантов. Для подтверждения диагноза аутосомно-рецессивного заболевания необходимо выявление патогенных/вероятно патогенных вариантов на обеих хромосомах в гомозиготном или компаунд-гетерозиготном состоянии.
Доброкачественные варианты - отличия в последовательности оснований ДНК в геноме (или в другой сравниваемой последовательности) разных индивидов или между гомологичными участками гомологичных хромосом индивида. Это изменение нуклеотидной последовательности, не являющееся патогенетической причиной исследуемого моногенного заболевания. Доброкачественные варианты нуклеотидной последовательности являются основой внутри- и межвидового разнообразия. При выявлении варианта неопределенного значения необходимо проведение дополнительных исследований для определения его связи с заболеванием: популяционного анализа частот замены, исследования сегрегации данной замены в семье, функционального анализа замены с использованием биоинформационных методов и/или модельных систем.
Все детектированные варианты указываются в заключении в соответствии с международной номенклатурой (http://varnomen.hgvs.org/). Изменение может быть записано с использованием геномной последовательности: chr10:g.90770499C>T (hg19) - в позиции 90770499 хромосомы 10 сборки генома 19 произошла замена цитозина на тимин; последовательности кодирующей ДНК: NM_005051.2:c23G>C - в23 положении кодирующей ДНК транскрипта NM_005051.2 гена произошла замена гуанина на цитозин или ENST00 000361445.4:c92_94del - в положении 92-94 кодирующей ДНК транскрипта ENST00000361445.4 произошло выпадение трех нуклеотидов. Если обнаруженный вариант приводит к аминокислотной замене в белковой последовательности, изменение может быть записано с использованием последовательности протеина: p.Trp26Cys - в положении 26 протеина произошла замена аминокислоты триптофан на цистеин. В некоторых случаях для удобства генетиков название мутации в соответствии с номенклатурой может быть продублировано традиционным названием данной мутации. Для обозначения цис/транс-положения выявленных вариантов применяются следующие символы: NP_003997.1:p.[Ser68Arg;Asn594del] - аллель содержит два разных варианта белковой последовательности, т.е. аллели находятся в цис-положении; NP_003997.1:p.[Ser68Arg];[Ser68=] - один аллель содержит вариант белковой последовательности, а второй аллель дикого типа; NP_003997.1:p.(Ser68Arg)(;)(Asn594del) - выявлено два варианта белковой последовательности, но неизвестно, в цисили транс-положении они находятся; NP_003997.1:p.[Ser68Arg];[Asn594del] - выявлено два варианта белковой последовательности в компаунд-гетерозиготном состоянии; NP_003997.1:p.(Ser68Arg) (;)(Ser68Arg) - вероятно гомозиготное состояние варианта последовательности белка, однако нельзя исключить компаунд-гетерозиготное состояние с протяженной делецией.
При отсутствии изменений нуклеотидной последовательности невозможно исключить генетическую природу заболевания у пробанда, так как: 1) в гене может быть мутация, не детектируемая использованным методом или не попавшая в систему детектируемых наиболее частых мутаций; 2) мутация может локализоваться в далеких от кодирующих последовательностей неисследованных регуляторных областях гена вследствие генетической гетерогенности наследственных болезней; 3) мутация, ответственная за данный клинический фенотип, локализуется в другом гене.
Общие рекомендации при проведении молекулярно-генетической диагностики
При направлении больных на молекулярно-генетическую диагностику необходимо учитывать, что это достаточно сложное и дорогостоящее исследование. Генетическая гетерогенность наследственных болезней диктует необходимость сбора как можно более полной клинической информации с целью выработки оптимальной последовательности исследования генов в каждой конкретной семье. До назначения диагностики желательно провести максимально возможное обследование пробанда с использованием доступных физикальных, лабораторных и инструментальных методов. Необходимо собрать подробную родословную семьи и при необходимости проведения дополнительных тестов предоставить материал родственников пробанда.
Материалом для проведения ДНК-анализа может быть любая ткань человека, содержащая ДНК. Наилучшим материалом для исследования является цельная кровь, собранная в пробирку с антикоагулянтом этилендиаминотетраацетатом, для большинства тестов допустимо использование сухих пятен крови на фильтровальной бумаге. В случае если больной член семьи недоступен, возможно проведение анализа с использованием различных биологических тканей после предварительного согласования с лабораторией. Материал для диагностики, как правило, не требует особых условий хранения и транспортировки, при температуре +4 °С жидкая кровь хранится в течение 2 нед, высушенные пятна крови и замороженная кровь могут храниться более 5 лет. ПД может проводиться на любом материале плода: ворсинах хориона, амниотической жидкости, пуповинной крови - выбор обусловлен только сроками беременности и возможностью проведения инвазивной процедуры.
В последнее десятилетие технологии секвенирования нового поколения (NGS) стали неотъемлемой частью медицинских генетических исследований по всему миру. В число лидеров в производстве оборудования и реагентов для NGS в 2018 году вошла китайская компания MGI, представившая линейку инновационных секвенаторов DNBSEQ®.
Запатентованная технология линейной клональной амплификации библиотек без этапа ПЦР позволяет исключить накопление ошибок ДНК-полимеразы и снизить риск количественного искажения библиотек. Готовые библиотеки представляют собой клубки из повторяющихся участков одноцепочечной ДНК (DNA Nano Ball, DNB).
Технология секвенирования MGI coolMPS® является новейшей разработкой в сфере NGS. В данном подходе используются нативные нуклеотиды и флуоресцентно меченные нуклеотид-специфичные моноклональные антитела. Использование антител позволяет в разы увеличить интенсивность сигнала и существенно повысить чувствительность приборов.
Разработка принципиально новых подходов в NGS и конкуренция между производителями обеспечивают стремительный прогресс технологий, упрощение биоинформатического анализа и снижение стоимости данных. Благодаря этому есть надежда, что в скором времени NGS-исследования - секвенирование генома, экзома, НИПТ, ПГД и другие востребованные типы исследований - станут доступны широкому кругу пациентов.
На правах рекламы
Заключение
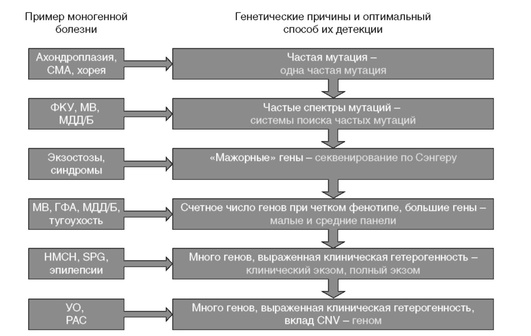
На сегодняшний день возможно проведение ДНК-диагностики практически любого заболевания, для которого известен ген. В РФ разработаны диагностические протоколы поиска мутаций различных генов более чем 400 моногенных болезней, поводится исследование таргетных панелей, экзомов и геномов. Задача консультирующего врача сегодня очень сложна - необходимо не только поставить клинический диагноз, но и определить оптимальную последовательность молекулярно-генетических анализов. Любой метод, применяемый в современной ДНК-диагностике, имеет ограничения по типу детектируемых мутаций. Алгоритм выбора методики молекулярно-генетического анализа в зависимости от генетической гетерогенности и преобладающего типа генетических вариантов представлен на рис. 15-6.
Для поиска вариантов, являющихся причиной болезни, а также для оценки их патогенности в современную практику медицинской генетики внедряются различные методы анализа РНК, методы функционального анализа и анализа экспрессии генов.
Только выявление непосредственной причины болезни - мутации в гене - дает возможность подтвердить диагноз на молекулярном уровне, определить риски рождения больного ребенка, провести диагностику носительства в семье и ПД.
Список литературы
-
Клиническая генетика: Учебник для мед. вузов / Под общ. ред. Н.П. Бочкова. М.: ГЭОТАР-Медиа, 2011. 537 с.
-
Кондрашева Е.А., Островский А.Ю., Юрасов В.В. Инвитро диагностика. Лабораторная диагностика. М.: Медиздат, 2007. 560 с.
-
Молекулярная клиническая диагностика / Под общ. ред. С. Херрингтон. М.: Мир, 1999. 558 с.
-
ПЦР «в реальном времени» / Под ред. Д.В. Ребрикова. М.: Бином, 2009. 215 с.
-
Рыжкова О.П., Кардымон О.Л., Прохорчук Е.Б. и др. Руководство по интерпретации данных последовательности ДНК человека, полученных методами массового параллельного секвенирования (MPS) (редакция 2018, версия 2) // Медицинская генетика 2019. Т. 18, № 2. С. 3-23. DOI: 10.25557/2073-7998.2019.02.3-23.
-
Свердлов Е.Д. Взгляд на жизнь через окно генома. В 3 т. М.: Наука, 2009.
-
Краснопольская К.Д. Наследственные болезни обмена. М.: Медицина, 2005.
-
Ребриков Д.В., Коростин Д.О., Шубина Е.С., Ильинский В.В. NGS высокопроизводительное секвенирование. 2-е изд. М.: БИНОМ. Лаборатория знаний, 2015. 232 с.
-
Grody W.W., Seligson D.B., Telatar M. Diagnostic Molecular Genetics in Emery and Rimons Principles and Practice of Medical Genetics. Churchill Livingstone, 2002. P. 723-751.
-
Sambrook J., Russell D.W. Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press, 2001. 2344 p.