Медицинская генетика : национальное руководство / под ред. Е. К. Гинтера, В. П. Пузырева, С. И. Куцева. - Москва : ГЭОТАР-Медиа, 2022. - 896 с. (Серия "Национальные руководства") - ISBN 978-5-9704-6307-9 |
Аннотация
Настоящее издание подготовлено коллективом авторов, абсолютное большинство которых хорошо известны не только отечественному, но и зарубежному читателю.
В книге представлены основные разделы генетики человека и медицинской генетики, такие как генетика развития человека, эпигенетика, генетика соматических клеток, популяционная генетика, фармакогенетика, иммуногенетика и система генов HLA. Также описаны современные представления об основных классах наследственных болезней: моногенных, включая популяционную генетику моногенных заболеваний, наследственных болезнях обмена веществ, митохондриальных заболеваниях, патологиях, обусловленных расширением зоны тандемных тринуклеотидных повторов, хромосомных болезнях, включая те, что обусловлены микрохромосомными перестройками, заболеваниях, связанных с геномным импринтингом, многофакторных болезнях, врожденных пороках развития и тератогенных синдромах.
Отдельная часть руководства посвящена лечению и профилактике наследственных болезней, а также этическим проблемам медицинской генетики. В ней подробно рассмотрена проблема генной терапии, в том числе с использованием методов редактирования генома, которые многими исследователями рассматриваются как наиболее перспективные в генотерапии заболеваний человека.
Книга будет полезна врачам практически всех специальностей с учетом все возрастающего вклада генетики в современную медицину, без которого невозможно ее дальнейшее совершенствование.
Глава 18. Моногенные наследственные болезни в популяциях человека
Моногенная наследственная патология характеризуется широким разнообразием и представлена описанием большого числа клинических и генетических форм. В настоящее время в мировой литературе описано более 25 400 различных фенотипов, из которых примерно 7000-8000 приходится на наследственные заболевания и синдромы [http://www.ncbi.nlm.nih.gov/OMIM]. Большая часть наследственных болезней встречается в популяциях человека довольно редко (с частотой <1:100 000), и многие заболевания представлены лишь единичными семьями. Очевидно, что необходимо расширение знаний об этиологии, включая молекулярно-гене-тическую природу наследственных болезней, их патогенезе, частоте и структуре наследственных заболеваний и особенностях распространения как всей генетически обусловленной патологии в целом, так и отдельных нозологических форм, что поможет разработать профилактические, лечебные и реабилитационные мероприятия.
Изучение груза наследственных болезней в популяциях человека, его количественных характеристик является важным как с теоретической, так и с практической точек зрения. Теоретическая сторона этих исследований определяется тем, что наследственные болезни составляют часть генетического груза человека. Определение величины груза наследственных заболеваний в популяциях человека является достаточно сложной и трудоемкой задачей, однако в этом есть реальная необходимость. Прежде всего, и это основное, изучение величины и структуры груза наследственных болезней позволяет определить объем медицинской, социальной, лечебной и реабилитационной помощи населению. Получение таких оценок дает возможность определить приоритеты развития некоторых служб здравоохранения, включая медико-генетическую, инициировать направления фундаментальных научных исследований и оценить в итоге результаты мероприятий по лечению, реабилитации и профилактике генетически детерминированной патологии.
Груз наследственных болезней и методы его выявления в современных популяциях человека
Одной из проблем современной медицины является груз заболеваемости человека, который продолжает расти с каждым годом, несмотря на совершенствование методов клинической и лабораторной диагностики. В «Бюллетене» Всемирной организации здравоохранения (ВОЗ) регулярно освещаются исследования по проблеме «Груз болезней в мире». Э.А. Мерфи и Г.А. Чейз (1979) справедливо отметили: «…груз существует как реальность, даже если современная наука не может пока его измерить». Неотъемлемой частью общего груза различных заболеваний человека, несомненно, является груз наследственных болезней.
Дж. Холдейн был первым, кто привлек внимание к существованию генетического груза, а сам термин был предложен в конце сороковых годов Х. Дж. Меллером (Muller H.J., 1950). В настоящее время выделяют множество различных видов генетического груза, однако наибольшее значение до сих пор имеют сегрегационный и мутационный грузы. Сегрегационный груз представляет собой разность между приспособленностью гетерозигот и средней приспособленностью популяции (Алтухов Ю.П., 2003). Мутационный груз - эта та доля общего генетического груза, которая возникает за счет мутантных аллелей, создается и поддерживается в результате баланса между давлением повторных мутаций и элиминацией их из популяции естественным отбором.
В медицинской генетике большое значение имеет оценка груза наследственных болезней, основанная на непосредственном медицинском обследовании населения. При достаточной генетической информированности всех врачей различных специальностей оценка генетического груза была бы возможна для многих популяций. Однако проблема груза наследственных болезней в популяциях человека, имеющая важное научное и прикладное значение, до сих пор остается недостаточно изученной, несмотря на то, что частота «патологических» генов в популяциях достаточно высока. В условиях улучшения медицинской и социальной помощи во многих странах больные с наследственной патологией будут не только дольше жить, что предполагает повышение генетического груза популяций, но и передавать мутации из поколения в поколение успешнее, чем это происходило ранее.
В середине 50-х годов XX в. появилось новое научное направление, позволяющее получить ответы на многие вопросы, связанные с оценкой генетического груза и разнообразия наследственных болезней в популяциях человека. На разных этапах развития с расширением знаний и получением новых результатов название данного направления менялось, модифицировалось, совершенствовалось, а именно: эпидемиологическая генетика (1959), генетическая эпидемиология (1979), популяционная геногеография наследственных болезней (1982), клиническая популяционная генетика, эпидемиология наследственных болезней (1984). Однако, независимо от названия на всех этапах развития, данное направление предусматривает, наряду с оценкой распространенности отдельных заболеваний и груза в целом, выявление механизма формирования груза моногенных наследственных болезней в популяциях человека. Для наследственной патологии эти принципы должны отличаться от уже разработанных в эпидемиологии инфекционных заболеваний и основываться на генетических закономерностях возникновения и распространения мутантных генов в популяциях и отдельных семьях.
Сложность и многогранность изучаемой проблемы обусловили основные направления исследований: определение качественных и количественных характеристик груза и разнообразия наследственной патологии и установление механизмов его формирования в различных популяциях человека. В результате генетико-эпидемиологических исследований клиническая медицина получает представление о распространенности, вариабельности проявления и спектре наследственной патологии у отдельных популяций, так как здравоохранению небезразлично появление накопления отдельных наследственных заболеваний в регионах, что часто приводит к социальным и медицинским проблемам. Популяционная генетика приобретает новые знания о характере генетической структуры данной популяции, которая наилучшим образом описывает территориальное распределение мутантных генов, определяет механизмы и особенности генетической дифференциации популяции по таким генам. Именно комплексный анализ предполагает разработку оптимальных профилактических мероприятий по контролю наследственных болезней в популяциях человека.
Для анализа груза наследственных болезней в медицинской генетике используют три основных подхода:
В различных популяциях человека выполнено большое количество работ, посвященных оценке груза наследственных болезней. Большинство работ реализовано с использованием первого подхода, что позволяет получить детальную качественную и количественную картину встречаемости исследуемой патологии в обследованном регионе. В последнее время при проведении генетико-эпидемиологических исследований все чаще применяются молекулярно-генетические методы, позволяющие определить не только частоту наследственной патологии, но и ее молекулярную природу.
Молекулярно-генетический анализ мутантных генов использовался при изучении цистиноза и миопатии Дюшенна во Франции и Японии, миотонической дистрофии в Квебеке, торзионной дистонии у евреев ашкенази, хореи Гентингтона в Уэльсе и Северной Ирландии, гемофилии в Швеции, Южной Австралии и Болгарии, атаксии-телеангиэктазии в Соединенных Штатах, атаксии и параплегии в провинции Торино, поясно-конечностной мышечной дистрофии в Японии, туберозного склероза в Таиланде, пигментного ретинита и синдрома Ушера в Норвегии, наследственной глазной патологии в Китае и Ньюфаундленде, буллезного эпидермолиза в Финляндии и Турции, адренолейкодистрофии в Австралии, гипопаратиреоза в США, наследственных болезней нервной системы в Мордовии, Владимирской, Волгоградской областях, Республике Башкортостан и т.д.
В последние 45 лет в различных странах мира получили распространение скринирующие программы у новорожденных с целью выявления наследственных болезней обмена веществ, большинство из которых наследуются аутосомно-рецессивно. Такие исследования позволяют получить детальное представление о частоте встречаемости данной патологии и гомо- и гетерозигот по мутантному гену в популяции. Основная цель скринирующих программ - выявление заболевания в доклинической стадии, когда возможно эффективное лечение. Во многих странах разработаны протоколы неонатального скрининга, учитывающие региональную частоту наследственной патологии.
ВОЗ в 1972 г. было рекомендовано создание регистров семей с наследственными заболеваниями для расширения на их основе объема генетического консультирования, профилактических программ и реабилитационных мероприятий в отношении пораженных членов семей.
Первой работой, посвященной изучению груза наследственных и врожденных заболеваний среди новорожденных, был регистр, созданный в Северной Ирландии (Stevenson A.C., 1959). Использовался обзорный метод получения данных: одномоментно от всех практикующих врачей. В результате функционирования регистра были получены сведения о больных с предположительно наследственной патологией в популяции Северной Ирландии. В этой работе оценивалась распространенность, а не частота наследственных болезней. Тем не менее значения распространенности аутосомно-доминантной и аутосомно-рецессивной патологии по Стивенсону были достаточно высокими. В то же время анализ этой работы показал ряд недостатков: были пропущены наиболее частые наследственные заболевания, встречающиеся у взрослого населения, а также занижены частоты некоторых аутосомно-рецессивных заболеваний при завышенном уровне отяго-щенности аутосомно-доминантной патологией. Кроме того, значительная часть заболеваний, которые Стивенсон относил к аутосомно-доминантным, таковыми в настоящее время не считаются, а многие состояния, включенные в перечень для регистрации, лишь условно можно считать патологическими.
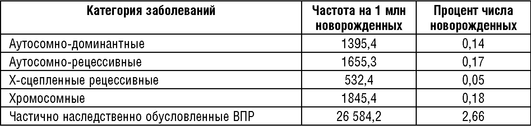
Следующее большое исследование по созданию регистра наследственных болезней было начато в Британской Колумбии (Канада) в 1953 г. (Trimble B.R., Smith M.E., 1977; Baird P.A. et al., 1988). Регистр функционирует так, что в него «стекаются» все данные о больных с диагностированной наследственной патологией из клиник, от частнопрактикующих врачей, учреждений социального обеспечения и т.д. (60 источников регистрации), а также статистические данные о численности населения, его рождаемости, смертности, движении населения провинции. Диагнозы классифицируются согласно МКБ-10. В результате объединения всех этих данных появляется возможность оценить как частоту отдельных наследственных болезней, так и групп заболеваний, а также всех наследственных заболеваний в целом. Эта частота для некоторых наследственных болезней может быть недооценена в связи с тем, что регистр существует чуть более 60 лет, а некоторые аутосомно-доминант-ные болезни проявляются позже. Но в остальных отношениях это лучшее из того, что есть в мире о частоте наследственных болезней в популяциях человека. Регистр включает в себя данные об отягощенности населения аутосомно-доминантными, аутосомно-рецессивными, Х-сцепленными заболеваниями, а также данные о ВПР, хромосомных заболеваниях и заболеваниях неизвестной этиологии (табл. 18-1).
В настоящее время успешно работают с регистрами в Великобритании, США, Франции, Швеции, ЮАР и во многих других странах. Наибольшее число регистров создано в США. В университете Юты функционирует генетическая база данных HGDBMS, состоящая из 6 модулей: демографической и генеалогической информации, данных о генотипах, клинических данных, результатов анализов крови, административного управления. Один из регистров функционирует в Алабамском центре медико-генетического консультирования. В Кливлендском генетическом центре для проведения клинико-генетических и генетико-эпидемиологических исследований с 1982 г. используется база данных GENE.SERV. Она содержит демографические и лабораторные (включая цитогенетические) данные, характеристики фенотипа, диагнозы и результаты консультаций, исходы. В генетическом центре штата Индиана (США) функционирует система MEGADATS, разработанная для планирования, совершенствования и оценки эффективности медико-генетической помощи, изучения генетики врожденных пороков, картирования хромосом и других целей.
Генетические регистры и базы данных с каждым годом приобретают все большее значение для всех областей здравоохранения. Активно функционируют:
Национальная генетическая база данных Израиля [http://server.goldenhelix.org/israeli; Zlotogora J., 2017]; проект «Геном Нидерландов» (GoNL) [http://www.nlgenome.nl], регистр финских болезней [http://www.findis.org/].
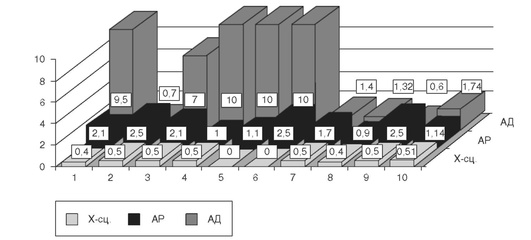
На рис. 18-1 представлены оценки частоты менделирующих наследственных заболеваний в популяциях человека, полученные разными авторами или коллективами авторов. В большинстве случаев эти оценки выведены путем суммирования данных из разных статей, посвященных эпидемиологии отдельных наследственных болезней в разных популяциях мира.
Проведение полноценного сравнительного анализа груза наследственной патологии в различных популяциях сопряжено с определенными трудностями, что обусловлено разницей в методологических подходах к ее изучению у различных авторов, а также количеством и качеством первичного материала. В известных исследованиях большинства авторов приводятся значения частот наследственных болезней, рассчитанные на новорожденных. Однако при проведении широкомасштабных работ по оценке большого числа нозологических форм приходится оперировать значениями распространенности или частоты встречаемости заболевания. О различии методологических подходов отечественных и зарубежных исследователей к оценке груза менделирующей наследственной патологии отчетливо свидетельствуют полученные результаты (см. рис. 18-1). Анализ их свидетельствует о том, что наибольшим колебаниям подвержена оценка частоты аутосомно-доминантных заболеваний, которые, по данным разных авторов, различаются больше чем на порядок.
Частота аутосомно-рецессивных заболеваний колеблется всего в два раза, что, скорее всего, связано с более строгими критериями отбора, а также с тем, что среди них редко встречаются состояния, которые можно считать условно патологическими.
Оценки же Х-сцепленных заболеваний практически не различаются между популяциями. В любом случае можно утверждать, что для получения оценок распространенности или частоты наследственных болезней в популяции материал должен быть определенным образом организован.
Наконец, еще одной формой проведения популяционно-эпидемиологических исследований является «обзорный» подход к анализу распространенности широкого спектра наследственных болезней. Преимущества данного метода заключаются в том, что наряду с решением ряда практических задач, таких как установление величины груза наследственной патологии и его качественной характеристики, он дает возможность подойти к проблеме мониторинга, изучению мутационного и этногене-тического процессов в популяциях человека (Гинтер Е.К., Зинченко Р.А. и др., 2006).
Оценка груза наследственных заболеваний в российских популяциях
Наиболее полноценным методом популяционно-генетических исследований, который позволяет провести обследование определенной популяции и выявить максимально возможное число нозологических форм наследственных заболеваний, является обзорный метод. Преимущества этого метода заключаются в том, что он дает возможность оценить величину груза наследственной патологии, провести анализ нозологического спектра, определить наиболее распространенные заболевания и основные популяционные механизмы их распространения. Кроме того, на основании данного подхода возможно создание базы для мониторинга наследственной патологии, изучение мутационного процесса в популяциях человека, описание генетического разнообразия, поддерживаемого в популяциях. Полученные в ходе исследования результаты являются основанием для улучшения работы региональной медико-генетической консультации, основой для создания региональных регистров и внедрения системы мониторинга за динамикой генетического груза. Помимо этого, эти исследования дают возможность оказать непосредственную помощь больным с наследственной патологией и их семьям.
Комплексные исследования по эпидемиологии наследственных болезней с применением обзорного метода начаты лабораторией генетической эпидемиологии Федерального государственного бюджетного научного учреждения «Медико-генетический научный центр им. Академика Н.П. Бочкова» (ИМГ АМН СССР) в популяциях бывшего СССР более 30 лет назад (Гинтер Е.К., 2006). Исследования проводятся по разработанному в лаборатории протоколу исследований.
Протокол исследований популяций включает изучение генетической структуры популяции через различные генетические системы: гены моногенных наследственных болезней (аутосомно-доминантные, аутосомно-рецессивные, Х-сцепленные), стандартные статистики популяционного анализа (F-статистики, демографические характеристики, индекс эндогамии, параметры изоляции расстоянием Малеко и др.) и ДНК-полиморфизмы ядерного генома. Сбор и обработка материала по трем направлениям проводятся одновременно в изучаемой популяции. На последнем этапе все данные, полученные при исследовании популяции по различным генетическим системам, обрабатываются и анализируются. Именно последний этап дает возможность понять основные микроэволюционные процессы, происходящие в популяции, влияющие на формирование груза и спектра моногенных наследственных заболеваний.
Наиболее сложным и трудоемким является сбор материала для медико-генетического исследования. Этот раздел протокола состоит из трех последовательных этапов (Гинтер Е.К., Зинченко Р.А. и др., 2006; Зинченко Р.А. и др., 2019).
На первом этапе определяются источники, через которые регистрируются данные о больных с предположительно наследственной патологией и создается единая база данных.
Первые исследования проводились в популяциях республик Средней Азии, где единственным источником регистрации служила разработанная в лаборатории специальная форма-анкета, распространяемая среди медицинских работников обследуемых районов. В анкете перечислены легко выявляемые симптомы наследственных болезней, распределенные в соответствии с основной классификацией по органному и системному типам заболевания (неврология, офтальмология, ортопедия, дерматология, оториноларингология, эндокринология, гематология), имеющие выраженное внешнее клиническое проявление. Все симптомы, перечисленные в карте-анкете, как отдельные наследственные заболевания, так и в сочетании с другими симптомами, позволяют выявлять целые группы заболеваний и множество наследственных синдромов, что существенно расширяет диагностические возможности исследований в выявлении не только моногенных заболеваний, но и ВПР и хромосомной патологии. Для примера: такой симптом, как тугоухость, может быть как изолированным широко гетерогенным заболеванием (описано более 100 генов, ассоциированных с изолированной тугоухостью), так и входить в перечень клинических симптомов наследственного синдрома - известно более 400 синдромов с различной степенью тугоухости.
Указанные в анкете симптомы позволяют не только выявить максимально возможное количество наследственных заболеваний из известных на настоящий момент, но и регистрировать новые, ранее не описанные формы, как частые, так и редкие, с разными типами наследования, в пропорции, близкой к той, которая есть в каталоге OMIM [http://www.ncbi.nlm.nih.gov/Omim/]. С учетом симптомов, перечисленных в анкете, определены врачи-специалисты, принимающие участие в осмотре больных и диагностике наследственной патологии в полевых условиях: генетик, педиатр, невролог, офтальмолог, ортопед, ЛОР-врач [1], дерматолог.
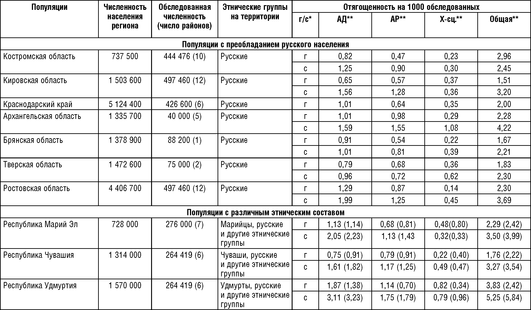
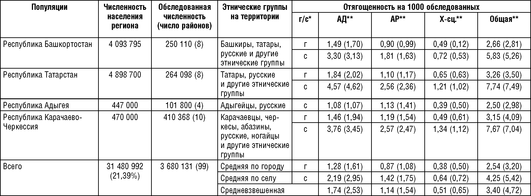
Проведение исследований в европейской части России ФГБНЦ «МГНЦ» началось более 35 лет назад несколько по другой схеме, хотя общая идея и содержание анкеты оставались постоянными на протяжении всех исследований. Регистрация семей с предположительно наследственной патологией велась через одного пораженного (в Средней Азии учитывались только семейные случаи, когда пораженных было не менее 2 в семье), и ввиду маленького размера семьи использовались многочисленные вторичные источники регистрации больных. К дополнительным источникам регистрации, используемым в работе, относятся: документы медико-социальной экспертизы (МСЭ), данные об инвалидах с детства, информация из домов-интернатов для слепых и слабовидящих, глухих и слабослышащих. Информация из всех источников информации объединяется в единой базе данных, что предполагает возможность регистрации одного и того же пациента из различных источников. Анализ работы показал, что анкета лучше «работает» в сельской местности, выявляя более 80% всех больных. В городах медицинский персонал хуже информирован обо всех больных на своем участке, и необходимость в использовании дополнительных источников регистрации более актуальна. В большинстве случаев для установления диагноза использованы молекулярно-генетические, биохимические, рентгенологические, электромиографические и другие методы исследований. Результаты эпидемиологических исследований, проведенных ФГБНУ «МГНЦ» в европейской части России, представлены в табл. 18-2.
Необходимо отметить, что единая методология, используемая в генетико-эпидемиологических исследованиях ФГБНУ «МГНЦ», позволяет сравнивать отягощенность населения разных популяций европейской части России наследственной патологией. Как следует из табл. 18-2, к настоящему моменту обследовано население 99 районов общей численностью 3 680 131 человек. При выборе районов для обследования учитывались история формирования популяции, миграции населения, этнографическое деление (в случае принадлежности популяции к определенной этнической группе). Полученные результаты изучения нескольких районов региона можно, как мы полагаем, экстраполировать на необследованные территории региона. В этом случае можно считать, что к настоящему моменту получены данные, дающие представление о распространенности наследственных болезней более чем у 21,39% населения России.
Окончание табл. 18.2
|
Примечание:
* г - малые города, райцентры районов; с - сельские субпопуляции;
** - в скобках указана отягощенность титульной нации региона, Х-сцепленная патология рассчитана на 1000 мужчин;
AP - аутосомно-рецессивный тип наследования;
АД - аутосомно-доминантный тип наследования.
При анализе отягощенности были выявлены следующие особенности. Самая высокая отягощенность населения во всех обследованных популяциях обусловлена аутосомно-доминантными, а самая низкая - Х-сцепленными заболеваниями. Нет достоверных различий между популяциями в их отягощенности Х-сцепленными болезнями. По крайней мере отчасти это может объясняться низкими абсолютными значениями отягощенности этой группы заболеваний. В сельских популяциях груз наследственных болезней достоверно выше, чем в городских, как для аутосомно-доминантных, так и для аутосомно-рецессивных заболеваний. Достоверные различия выявлены внутри групп сельских и городских популяций по отягощенности как аутосомно-доминантными, так и аутосом-но-рецессивными заболеваниями. Различия прослеживаются также при сравнении отягощенности основными видами наследственной патологии различных этнических групп.
Например, отягощенность аутосомно-доминантной и аутосомно-рецессивной патологией марийцев, чувашей, башкир, удмуртов, татар, карачаевцев, ногайцев, абазин и черкесов оказалась выше, чем русских и адыгейцев, как в городском, так и сельском населении. Суммарная отягощенность населения наследственными болезнями варьирует от 1:91 обследованных среди сельского населения ногайцев Республики Карачаево-Черкессия до 1:658 человек в городских популяциях Костромской области.
Важно подчеркнуть, что дифференциация по отягощенности аутосомными заболеваниями существует между популяциями разного иерархического уровня. Чем ниже иерархический уровень популяции, тем больше обнаружено различий между популяциями. Наибольшие различия наблюдались между сельскими администрациями в рамках одного района. Например, в Шарканском районе Удмуртской Республики груз в различных сельских администрациях примерно равной численности (500-1000 человек) варьировал от 0 до 16,34:1000 человек; в Кугарчинском районе Республики Башкортостан - от 0до 18:1000. Менее выражены различия в отягощенности между отдельными районами одного региона (например, вариация от 1,91 до 5,77 на 1000 человек между отягощен-ностью сельского населения двух соседних районов Удмуртской Республики; от 4,76 до 11,82 на 1000 человек между отягощенностью сельского населения двух соседних районов Республики Карачаево-Черкессия). И, наконец, наименьшие различия определены между обследованными субъектами Российской Федерации.
Анализ отягощенности наследственных болезней исследованных российских популяций показал, что существует отчетливая дифференциация по этому показателю между отдельными популяциями как внутри каждой из 14 обследованных территорий, так и между ними. Есть все основания полагать, что дифференциация исследованных популяций существует также по частотам генов наследственных болезней. Следует заметить, что полученные оценки отягощенности или распространенности наследственных болезней, особенно в сельских популяциях Кировской области, Республики Марий Эл и Чувашии, по своим абсолютным значениям очень близки к оценкам частоты наследственных болезней (по данным Регистра врожденной и наследственной патологии) в популяции Британской Колумбии (Канада).
Таким образом, можно отметить, что настоящий период характеризуется накоплением и систематизацией данных о распространенности наследственной патологии в различных популяциях, поисками адекватных подходов к оценке груза наследственной патологии и его динамики, попытками анализа различных факторов популяционной динамики в накоплении мутантных генов в популяциях человека.
Оценка разнообразия наследственной патологии в популяциях человека
Оценки разнообразия наследственных болезней в популяциях человека появились относительно недавно, и проведение полноценного сравнительного анализа по данной характеристике популяций является сложной задачей, что связано с несколькими основными причинами: различия в подходах выявления наследственных болезней в разных исследованиях; ограниченность фактически зарегистрированного среди населения материала о разнообразии наследственной патологии; выявленная генетическая гетерогенность большинства наследственных болезней при схожих клинических проявлениях. Количество и качество источников информации о больных с наследственной патологией существенно различаются в разных популяциях. У ряда авторов приводятся значения частот наследственных болезней, рассчитанные на новорожденных, причем информация о частотах отдельных заболеваний чаще представляет собой сводные данные, собранные многими авторами в отдельных популяциях, при этом анализируемые выборки отличаются. При проведении широкомасштабных работ по оценке большого числа нозологических форм обычно приходится оперировать значениями распространенности заболевания. В настоящее время в литературе имеется несколько публикаций, в которых обобщены результаты по частотам отдельных нозологических форм наследственных заболеваний и приведены сводные таблицы. Оценки частот наследственных заболеваний в европейских популяциях впервые представлены К. Картером в 1977 г. (табл. 18-3). Необходимо учитывать тот факт, что частоты, систематизированные и приведенные в таблице, получены разными авторами при использовании различных методов. К. Sankaranaraynan (1998) скорректировал приведенные К. Картером данные. В анализ также включены современные данные сайта «Orpha.net» [http://www.orpha.net/].
Окончание табл. 18.3
|
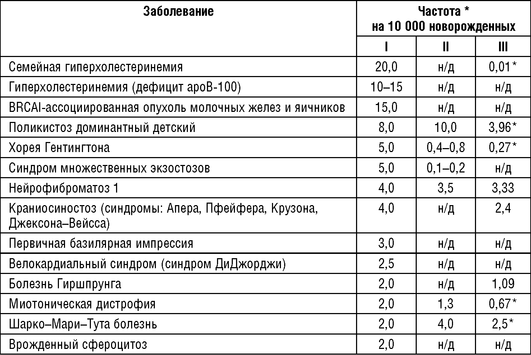
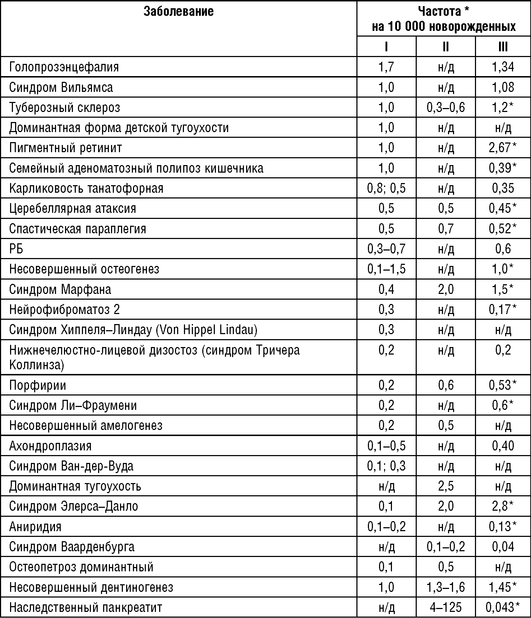
Примечание: н/д - нет данных; * - данные о распространенности заболевания; I - Carter C.O., 1977; II - Sankaranaraynan К., 1998; III - Orpha.net [http://www.orpha.net/, 2019].
Как следует из данных, приведенных в табл. 18-3, имеются существенные отличия в распространенности наследственных заболеваний, представленных указанными авторами. Возможно, это связано с отбором популяций, в которых наблюдается накопление отдельных заболеваний, что привело к некоторому завышению частот наследственных заболеваний.
В публикации K. Sankaranaraynan (1998) приведены данные только о частотах моногенных заболеваний с аутосомно-доминантным типом наследования. Эта группа заболеваний оказалась наиболее исследована в связи с изучением действия ионизирующего излучения и уточнением генетического риска в популяциях человека. Частоты некоторых заболеваний (табл. 18-3) в публикации К.Sankaranaraynan оказались выше, чем в сводных данных К. Картера и Orpha.net. Можно допустить, что это связано с улучшением диагностики и информированности врачей о наследственной патологии.
В табл. 18-4 приведены наиболее распространенные аутосомно-рецессивные заболевания по данным исследований, опубликованных К. Картером и Orpha.net. К наиболее частым наследственным заболеваниям в европейских популяциях относятся следующие: гемохроматоз, муковисцидоз, олигофрения, несиндромальная врожденная тугоухость, ФКУ, СМА, пигментные дистрофии сетчатки, поясно-конечностные миопатии, адреногенитальный синдром и серповидноклеточная анемия.
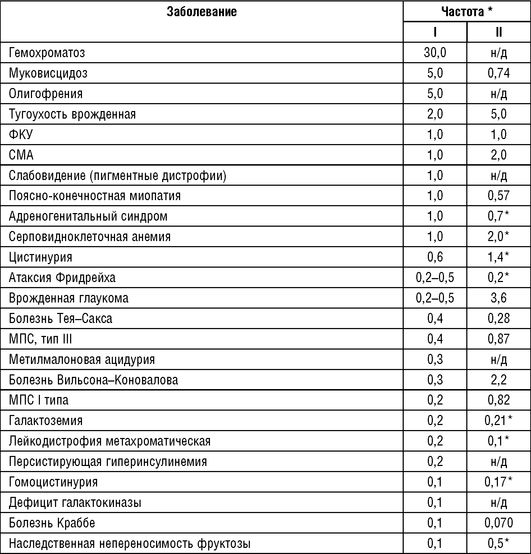
Примечание: н/д - нет данных; * - данные о распространенности заболевания; I - Carter C.O., 1977; II - Orpha.net [http://www.orpha.net/, 2019].
Частые в европейских популяциях Х-сцепленные заболевания приведены в табл. 18-5. В группу наиболее частых попадают прогрессирующая мышечная дистрофия Дюшенна, гемофилия А, ихтиоз Х-сцепленный и синдром ломкой Х-хромосомы.
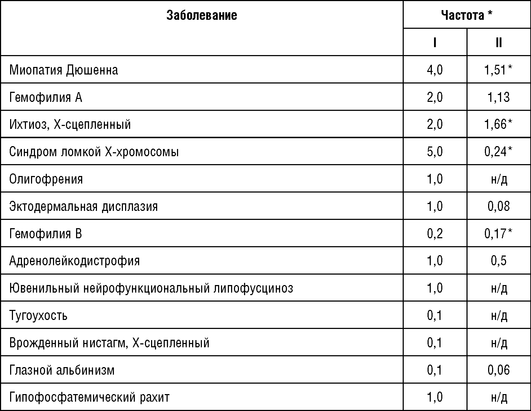
Примечание: н/д - нет данных; * - данные о распространенности заболевания; I - Carter C.O., 1977; II - Orpha.net [http://www.orpha.net/, 2019].
Oбращаeт на себя внимание отсутствие указаний на различные клинические типы наследственных заболеваний в приведенных публикациях. Скорее всего, это связано с особенностями получения информации автором из источников, в которых заболевания при регистрации кодируют по международной классификации болезней. В настоящее время в связи с картированием большого числа генов наследственных заболеваний, в том числе с идентичным клиническим проявлением, стало возможным выделение некоторых из них в самостоятельные нозологические формы.
Независимо от некоторых формальностей, список частых наследственных болезней, представленный К. Картером, а также в работах К. Sankaranaraynan и Orpha.net [http://www.orpha.net/, 2019], дает представление о том, какие наследственные болезни являются относительно частыми, а какие - более редкими. Oн оказывается также полезным для выявления накопления или, напротив, заметного уменьшения частоты определенных наследственных болезней в отдельных популяциях человека.
Необходимо отметить, что наиболее интересными являются результаты созданного в провинции Британская Колумбия (Канада) регистра наследственной патологии, функционирующего и в данное время. Анализ результатов его работы проведен Р.А. Baird (1988). Регистр включает сведения о частоте наследственной патологии у населения провинции в пересчете на 1 млн новорожденных, диагнозы классифицированы в соответствии с Международной классификацией болезней 10-го пересмотра (табл. 18-6).
Окончание табл. 18.6
|

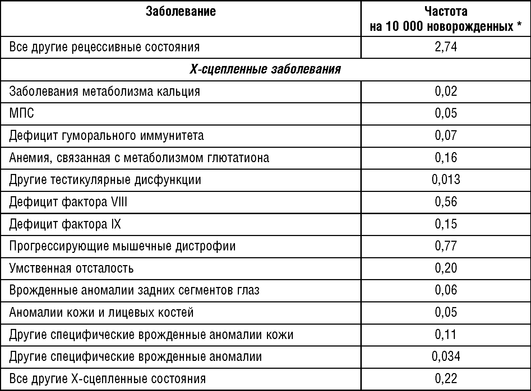
Примечание: * Х-сцепленная патология приводится на 10 000 новорожденных мальчиков.
Сравнительный анализ показал, что частоты большинства наследственных заболеваний по данным регистра в Британской Колумбии существенно ниже, чем в работах К. Картера и К. Sankaranaraynan. Oднако, несмотря на некоторые различия в интерпретации данных, результаты эпидемиологических исследований сопоставимы и могут рассматриваться как базисные.
В некоторых работах показано отличие частот отдельных нозологических форм в азиатских популяциях от европейских. В табл. 18-7 приведены частоты частых доминантных, рецессивных и Х-сцепленных заболеваний в Японии (Matsunaga E., 1986).
Окончание табл. 18.7
|
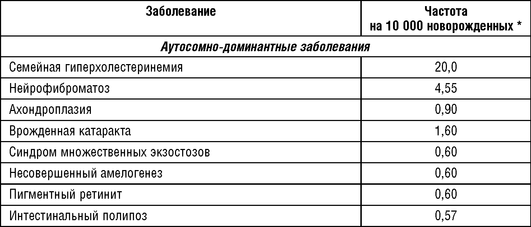
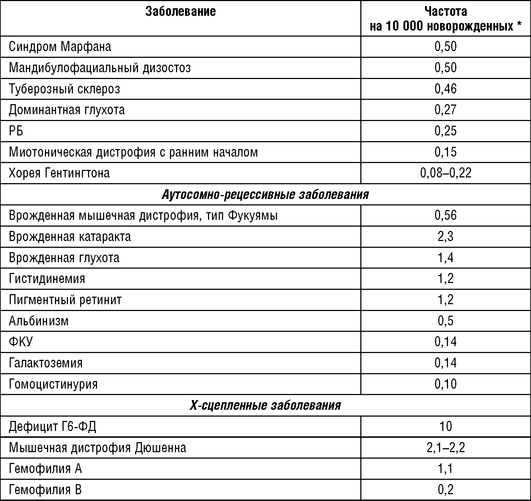
Как следует из табл. 18-7, спектр частых для Японии наследственных заболеваний в целом не отличается от спектра наследственных болезней в европейских популяциях и в популяции Британской Колумбии, однако отмечаются различия в частотах, и кроме того, выделены некоторые частые заболевания, характерные именно для данной популяции.
На данном этапе удалось в той или иной мере установить основную, наиболее стабильную часть нозологического спектра для некоторых российских популяций европейской части России на основании генетико-эпидемиологических исследований, которые приведены в табл. 18-8.

Окончание табл. 18.8 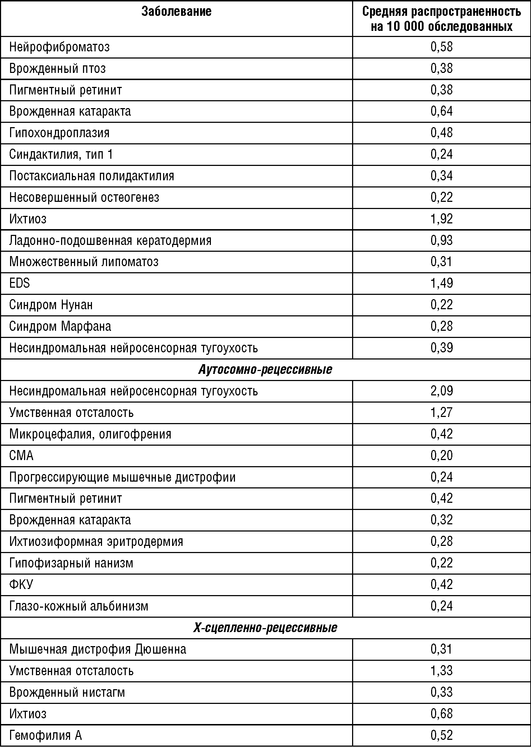
|
Примечание: * Х-сцепленная патология приводится на 10 000 обследованных мужчин.
Список частых наследственных заболеваний в европейской части России хорошо сопоставим со списком, приведенным для европейских популяций и для популяции Британской Колумбии.
Дифференциация по разнообразию и частотам менделирующих заболеваний между отдельными популяциями или этническими группами
Приведенные выше значения распространенности частых наследственных заболеваний не описывают особенности распределения отдельных заболеваний по популяциям или этническим группам. Oднако было бы неверным предполагать, что в современных популяциях человека наследственные болезни распространены равномерно и не наблюдается дифференциации как в самом нозологическом спектре, так и в частотах отдельных заболеваний между популяциями, народами. На протяжении почти всей эволюционной истории человека размеры его популяций были относительно малого размера. Человеческая популяция была разделена на множество изолированных, скрещивающихся внутри себя групп - «изолятов». До недавнего времени таких групп было много, некоторые из них существуют до сих пор. Численность их увеличивалась с различной скоростью, и большинство этих групп стали смешиваться относительно недавно, формируя свой, специфический, генофонд популяций. Невозможно точно определить, каким образом частоты генов наследственных болезней меняются под влиянием факторов современной цивилизации.
Среди направлений современной медицины и биологии популяционная генетика наследственных болезней занимает особое место, так как позволяет ответить на некоторые вопросы об эволюции популяций человека, их происхождении, оценить особенности генофонда, то есть совокупную наследственную информацию, которая передается от родителей к потомкам и сохраняется во времени в условиях нормально колеблющейся среды. Жизнь отдельной особи ограничена во времени, и ее генотип сохраняется постоянным в течение всей жизни. Популяция, в противоположность отдельной особи, практически бессмертна; она может быть большой или малой по численности, иметь обширный или ограниченный ареал и внезапно или постепенно изменяться по своему генетическому составу от поколения к поколению. Поэтому изучение генетики популяций неизбежно связано с изучением эволюции, которая с генетической точки зрения является ни чем иным, как процессом кумулятивного изменения наследственных особенностей видов. Другими словами, популяционная генетика наследственных болезней человека изучает поведение генов, обусловливающих эти заболевания в популяциях, и отвечает на ряд вопросов, в частности, как влияют на ее генетическую структуру такие факторы, как мутационный процесс, отбор, миграции, инбридинг, случайное изменение генных частот (Гинтер Е.К., 2003).
Природная популяция является элементарной единицей эволюционного процесса, а лучшее определение популяции принадлежит Н.В. Тимофееву-Ресовскому (1973), согласно которому «популяцией называется группа организмов, достаточно долго проживающая на определенной территории, в которой в большей или меньшей степени осуществляется панмиксия и которая отграничена от соседних таких же совокупностей особей данного вида той или иной степенью давления тех или иных форм изоляции». Именно популяция обеспечивает генетическую преемственность поколений, регуляцию таких биологически важных свойств, как плодовитость, численность, устойчивость к заболеваниям и, главное, протекание микроэволюционного процесса.
Частоты генов в популяции остаются неизменными при соблюдении основных условий выполнения закона Харди-Вайнберга. Oсновное утверждение закона Харди-Вайнберга, сформулированного в 1908 г, заключается в том, что в отсутствие действия на популяцию факторов популяционной динамики, таких как миграции, дрейф генов, мутационный процесс и естественный отбор, частоты генотипов и генов остаются неизменными в неограниченно долгом ряду поколений. Подобные идеальные популяции в природе практически не встречаются, так как всегда существуют факторы, сдвигающие их с точки равновесия, нарушающие ее стабильность, - случайный дрейф генов, мутации, миграции и естественный отбор. Каждый из этих факторов имеет количественную меру и может изменяться в широких пределах, но, так или иначе, все они оказывают влияние на любую природную популяцию. Oднако исследователи очень редко наблюдают в изучаемых ими популяциях отклонение в частотах генов от равновесного.
В популяции обычно действует одновременно несколько факторов популяционной динамики. Дарвин считал отбор основной движущей силой эволюции, так как он является единственным фактором, способным направленно изменять частоту генов в популяции. Oднако если в отношении нормальных признаков естественный отбор может как повышать, так и понижать частоту генов, определяющих эти признаки в популяции, то применительно к наследственным болезням почти всегда действует негативный отбор. Центральной концепцией отбора является дарвиновская приспособленность. Приспособленность генотипа определяется как способность дожить до репродуктивного возраста и оставить потомство, приспособленность оптимального генотипа принимается равной единице. В результате в зависимости от того, насколько сильно снижена приспособленность больных конкретным наследственным заболеванием, устанавливается равновесие между давлением вновь возникающих мутаций и давлением естественного отбора. Существенно, что в разных популяциях человека снижение приспособленности у больных одним и тем же наследственным заболеванием обычно одинаково, и поэтому уровень равновесия между давлением мутаций и отбора в них также одинаков. Если же в разных популяциях применяют методы лечения разной эффективности, частота заболевания в них может отличаться, однако эти различия будут несущественны. Следовательно, в большинстве случаев естественный отбор не может быть фактором, вызывающим дифференцировку популяций по частотам генов доминантных и Х-сцепленных наследственных болезней.
Несколько сложнее обстоит дело с рецессивными наследственными заболеваниями. В этом случае естественный отбор, безусловно, действует против всех гомозигот по генам рецессивных наследственных болезней. В то же время на гетерозиготных носителей мутантных генов, которых значительно больше, чем гомозиготных носителей, т.е. больных, отбор может действовать по-разному. Наиболее известные примеры действия отбора в пользу гетерозиготных носителей мутантных генов представляют собой гены β-талассемии и серповидноклеточной анемии. β-Талассемия и серповидноклеточная анемия распространены по всему субтропическому и тропическому поясу, в частности, в Средиземноморье. В условиях распространения в этих же регионах тропической малярии, которая выступает в качестве фактора отбора, гетерозиготные носители указанных мутантных генов, по-видимому, имеют селективное преимущество, и частота мутантных генов может возрастать, формируя систему сбалансированного полиморфизма. Для других рецессивных наследственных заболеваний, обнаруживающих достаточно высокую частоту в нескольких этнических группах или странах, значение естественного отбора в создании селективных преимуществ для носителей соответствующих мутантных генов не было доказано, хотя и предполагалось некоторыми авторами. Для большинства рецессивных наследственных заболеваний, которые встречаются в популяциях с низкими или очень низкими частотами, действие естественного отбора в пользу гетерозиготных носителей мутантного гена даже не предполагается. Более того, некоторые авторы полагают, что гетерозиготные носители мутантных генов могут иметь слегка сниженную по сравнению с носителями нормальных аллелей приспособленность.
В популяциях человека действие отбора в настоящее время ограничено в связи с успехами медицины, улучшением условий жизни и снижением смертности. Oднако он продолжает действовать, хотя может осуществляться только за счет избирательной репродуктивности и в основном в период внутриутробного развития. Вместе с тем уменьшение числа детей в семьях сужает сферу действия отбора. Этот отбор относится к категории стабилизирующего и устраняет вредные мутации в исторически сложившихся популяциях человека.
Равновесие генных частот в популяции предполагается при отсутствии миграций. Наряду с этим между реально существующими популяциями человека постоянно происходит миграционный процесс, который вносит дополнительную генетическую изменчивость в популяцию и может приводить к изменению генных частот. Как правило, миграции способствуют выравниванию генных частот в популяциях и сдерживают процесс дифференциации субпопуляций по частотам генов. Oднако в некоторых случаях миграция может приводить к возрастанию различий в частотах генов в двух популяциях, когда определенную часть мигрантов составляют группы родственных индивидуумов. Это легко себе представить, если вообразить, что новая популяция образуется за счет выселения из существующей популяции определенного клана или рода. Такое событие может привести к возникновению дифференциации исходной и вновь возникшей популяции по частотам генов.
Изучению миграций как фактора, изменяющего генетическую структуру популяций и, соответственно, генофонд популяций, посвящено большое число исследований. Так, H. Picheral (1985) в исследовании степени миграции французов начиная с XVI в. выявил, что большая часть миграций происходила на небольшие дистанции, так как формирование городов происходило за счет населения близлежащих районов. Сельские же популяции остаются относительно стабильными. В то же время формирование населения Парижа происходило за счет выходцев из самых разных районов Франции. Исследование, проведенное в Италии C.R. Guglielmino и соавт. (1998), показало, что за последние 50 лет действие урбанизации привело к нарушению репродуктивной изоляции в общинах малого размера.
Не может приводить к возникновению генетической дифференциации по генам наследственных болезней и мутационный процесс. Мутации являются одним из стохастических факторов, влияющих на равновесие генных частот в популяции. Мутации возникают постоянно и приводят к появлению новых аллелей в популяции, изменяя ее генетическую структуру. Oднако их сохранению противодействует естественный отбор, и в результате возникает равновесие между появлением новых мутаций и их элиминацией, что позволяет сохранять в популяции равновесные генные частоты. Средняя частота спонтанного возникновения мутаций у человека различна и находится в пределах от 1,0×10-5 до 6,08×10-6 на одну гамету за каждое поколение. Наследственные болезни, такие как хромосомные и некоторые аутосомно-доминантные, поддерживаются на определенном уровне в популяции исключительно за счет мутационного процесса. В человеческих популяциях, которые характеризуются медленной сменой поколений (продолжительность одного поколения в среднем составляет 25-30 лет), мутационное давление как фактор, определяющий эволюционные изменения, играет незначительную роль. Частота вновь возникающих мутаций на гамету на поколение - слишком незначительная величина, чтобы быть причиной генетической дифференциации популяций.
К факторам популяционной динамики относят также неслучайный инбридинг, обусловленный положительной брачной ассортативностью. Понятие «инбридинг» широко используется в популяционной генетике при описании особенностей генетической структуры популяций. Случайное скрещивание, или панмиксия, - это абстракция. В современных аутбредных популяциях скрещивание может быть почти случайным по таким признакам, как группы крови или варианты ферментов, однако по некоторым наследственным признакам, таким как тугоухость, они, конечно, не случайны. Менее очевидны, но более распространены браки, ассортативные по психологическим или социальным особенностям. Ассортативность ведет к увеличению доли гомозиготных и уменьшению доли гетерозиготных лиц в популяции, что теоретически должно нарушать равновесие частот генотипов и, соответственно, приводить к уменьшению внутрипопуляционной генетической изменчивости и увеличению межпопуляционной генетической изменчивости.
Oдин из типов ассортативных браков - брак между родственниками, или инбредный брак. По определению В.А. Ратнера (1977), инбридингом называют ограничение свободы скрещивания индивидуумов, связанное лишь отношениями родства, когда, независимо от генотипа, близкородственные особи имеют повышенную частоту скрещивания. Ч. Ли (1978) определяет инбридинг как коэффициент корреляции между гаметами g1 и g2 , при объединении которых образовалась особь z.
Инбридинг имеет количественную меру. Для оценки инбридинга в популяции чаще используются коэффициент инбридинга, введенный С. Райтом (1973), и коэффициент родства Дж. Малеко (1973). В качестве меры дифференциации популяции С. Райт предложил различать три статистики. FIS - локальный инбридинг - является мерой корреляции между объединяющимися гаметами в субпопуляции, позволяет оценить брачную ассортативность в популяции. FST - случайный инбридинг - корреляция между случайно объединяющимися гаметами в субпопуляции относительно всей популяции, используется для сравнения степени дифференцированности субпопуляций по частотам генов и является количественной мерой для измерения эффективности дрейфа. FIT - тотальный инбридинг - складывается из локального и случайного инбридинга. Различные определения термина «инбридинг», встречающиеся в литературе, не меняя смысла изучаемого параметра, отражают лишь способ формирования данной характеристики.
По данным BO3, численность инбредных популяций в мире составляет более 1 млрд человек. Формально инбридинг меняет частоты не генов, а генотипов, увеличивая доли гомозиготных индивидуумов и уменьшая соответственно доли гетерозиготных индивидуумов в популяции. Поэтому в популяциях, в которых распространены близкородственные браки, ожидается и нередко наблюдается большое число больных рецессивными наследственными заболеваниями.
Накопление разнообразной рецессивной патологии, обусловленное высоким уровнем неслучайного инбридинга, было выявлено во многих популяциях человека. Oбследование населения Кувейта выявило накопление ряда аутосомно-рецессивных заболеваний (Teebi A.S., Farag T.I., 1997). Исследования, проведенные в Японии, показали, что при средней частоте браков в популяции между двоюродными сибсами, равной 1%, частота этих браков среди родителей больных альбиносов составляет 25%, больных амаврозом - 53%, ихтиозом - 40%, пигментной ксеродермой - 26%. Работы из Карнатаки (Южная Индия) показали, что наиболее предпочтительными в этой популяции являются кровнородственные браки. Коэффициент инбридинга, рассчитанный для всей популяции, составил 0,0271. Oтягощенность менделирующей наследственной патологией, представленная в работе D.A. Ratha-Rama (1987), составила: аутосомно-доминантной - 2,7 на 1000 человек, аутосомно-рецессивной - 5,9 на 1000 человек, Х-сцепленной - 1,2 на 1000 мужчин. Oтмечен высокий удельный вес аутосомно-рецессивной патологии в общей отягощенности наследственными заболеваниями.
Еще одним фактором микроэволюции, теоретически приводящим к генетической дифференциации популяций по частотам генов наследственных болезней, может быть дрейф генов, или случайное изменение частоты гена в популяции при переходе к следующему поколению. Проявлениями эффективного дрейфа являются эффект родоначальника, обозначающий в строгом смысле преимущественное распространение в популяции генов одного из родоначальников, а также эффекты «горлышка бутылки» и популяционной волны, когда после резкого сокращения численности популяции, обусловленного теми или иными внешними причинами (войны, эпидемии и т.д.), следует быстрый рост популяции. При этом может случайным образом произойти фиксация даже тех генов, которые снижают приспособленность своих носителей. Случайная потеря и случайная фиксация - это крайние случаи; в промежуточном случае (что более характерно для современных популяций) в популяции сохраняются оба аллеля, но их частота флуктуирует случайным образом. Oднако если фиксация уже произошла, обратный процесс невозможен. Вероятность фиксации с увеличением числа поколений стремится к 1.
Изучение природных популяций показывает, что, как правило, они не представляют собой единой панмиксной единицы. В настоящее время практически принимается за догму: если в популяции не распространена брачная ассортативность, а кровнородственные браки встречаются не чаще, чем в панмиксной популяции, то весь инбридинг складывается только за счет подразделенности популяций (FST ), являющейся количественной мерой дрейфа генов. Таким образом, высокий инбридинг в популяциях человека может быть обусловлен как большим количеством кровнородственных браков (неслучайная составляющая инбридинга FIS ), так и географической и социальной подразделенностью (случайная составляющая инбридинга FST ).
Работы, посвященные разнообразию наследственных болезней в популяциях человека, в основном выполнены в изолятах, различных этнических группах, национальных и региональных подразделениях либо на малочисленных популяциях. Как правило, это небольшие группы людей, сохраняющие определенную традиционную культуру из поколения в поколение. На основании этих исследований удалось в той или иной мере установить разнообразие наследственных болезней в различных популяциях, определить наиболее часто встречающиеся заболевания, установить факторы возникновения и реализации патологических генов.
Oдной из первых, теперь уже классических работ по накоплению редких наследственных болезней в изолированных популяциях Северной Америки была работа В. Мак-Кьюсика в популяции амишей, меннонитов и гаттеритов. Эти популяции относятся к анабаптистскому движению, начавшемуся в Европе в XVI в. Наиболее полное медико-генетическое обследование было проведено В. Мак-Кьюсиком (1973) в популяции амишей Ветхого Завета. В течение 100 лет относительно небольшие по численности группы амишей иммигрировали из Западной Европы в США, образуя слабо связанные между собой общины в различных штатах. В последующие годы их численность возросла более чем в 8 раз, то есть наблюдался эффект «горлышка бутылки». Все общины оказались практически полностью изолированными от окружающего населения.
Несмотря на попытки избежать кровнородственных браков с двоюродными сибсами, коэффициент инбридинга достаточно высокий во всех группах амишей. Коэффициент случайного инбридинга составил 0,035, высокий уровень обусловлен строгой эндогамией в течение двух веков. Географическая подразделенность, исходно малый размер и популяционная волна объясняют накопление отдельных нозологических форм в элементарных популяциях амишей, как аутосомно-доми-нантных, так и аутосомно-рецессивных и Х-сцепленных. У амишей было описано несколько прежде неизвестных наследственных заболеваний, в том числе синдром гипоплазии хряща и волос, синдром Троера, синдром MAST и ряд других. В табл. 18-9 приводятся некоторые наследственные болезни, обнаруженные с высокими частотами в популяции амишей.
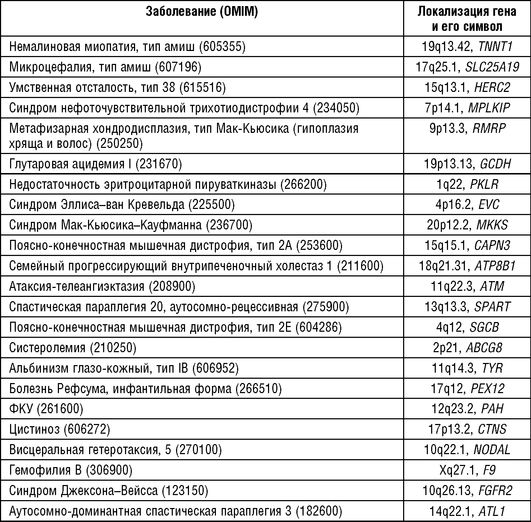
Для некоторых наследственных заболеваний у амишей удалось прямо продемонстрировать «эффект родоначальника», доказав, что все семьи с одним и тем же наследственным заболеванием объединяются в одну родословную с общими предками. Молекулярно-генетические исследования, проведенные в популяции амишей, также подтвердили предположение о роли эффекта родоначальника и дрейфа генов в распространении наследственных болезней.
Так, при синдроме Джексона-Вейсса, или синдроме краниостеноза, гипоплазии средней части лица и аномалии стоп, картирован ген FGFR2 в области 10q26.13, мутации в котором явились причиной развития заболевания. Oдна и та же мутация в гене тропонина Т1 (TNNT1) найдена у всех больных амишей с особой формой немалиновой миопатии, частота которой в этой популяции оценивается в 1:500. У амишей из графства Ланкастер (Пенсильвания), больных глутаровой ацидемией I типа, найдена только одна мутация в гене GCDH в гомозиготном состоянии. Своеобразный результат получен при изучении синдрома Эллисаван Кревельда, 50 случаев которого зарегистрировано в том же графстве, что и глутаровая ацидемия. У всех больных, относящихся к обширной родословной, найдена миссенс-мутация в гомозиготном состоянии, с заменой аргинина на глицин в 760 положении белка, контролируемого геном EVC. Позднее было показано, что эта мутация, скорее всего, представляет собой редкий полиморфизм в гене EVC. В то же время эта мутация подтверждает правильность предположения о роли эффекта основателя в распространении синдрома среди амишей (Ruiz-Perez V.L. еt al., 2000).
Изучение генетико-демографических характеристик популяций амишей, меннонитов и гаттеритов показало, что основные факторы популяционной динамики, влияющие на структуру отягощенности наследственными заболеваниями, одинаковы для этих популяций и обусловлены в основном дрейфом генов, однако роль других факторов популяционной динамики различна.
У амишей локальная приуроченность и семейное накопление редкой рецессивной патологии обусловлены «эффектом родоначальника», естественным отбором, дрейфом генов. Для гаттеритов характерны подразделенность, дрейф генов, естественный отбор, что, в свою очередь, привело к накоплению семейных случаев рецессивной и доминантной патологии. Для меннонитов: случайный инбридинг, «эффект основателя», дрейф генов. Изучение этих изолятов позволило проанализировать степень их генетической дифференциации от исходных популяций и оценить вклад параметров популяционной структуры в скорость эволюционного процесса.
На основании проведенных исследований у амишей и меннонитов при помощи метода секвенирования нового поколения разработаны профилактические скринирующие программы. Анализ секвенирования нового поколения разработан с использованием технологии множественной мультиплексной ПЦР (ArcherDX) для 162 различных генетических синдромов, ассоциированных с 202 патогенными генетическими вариантами. Программа скрининга может помочь снизить заболеваемость и смертность от этих заболеваний в группах высокого риска, а также снизить общие медицинские расходы и улучшить результаты лечения пациентов (Crowgey E.L. et al., 2019).
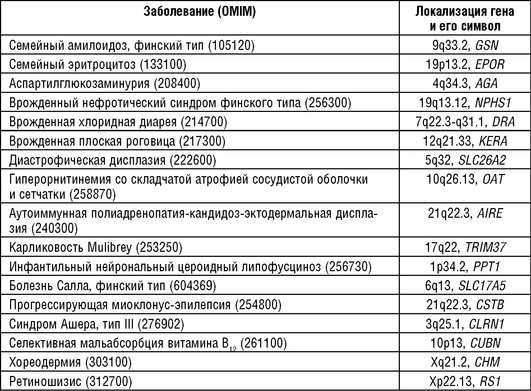
Исследования, проведенные в финской популяции, привели к обнаружению более 20 форм уникальной рецессивной патологии и 2 форм аутосомно-доминантной, которые очень редко выявляются в других странах, включая соседние скандинавские страны (Norio R. et al., 1973; Peltonen L. et al., 1997). К этим заболеваниям относятся аутосомно-доминантные амилоидоз, финский тип и эритроцитоз (40 больных). Среди рецессивных заболеваний - аспартилглюкозаминурия (частота в Финляндии 1:26 000); синдром эндокринопатии-кандидоза-эктодермальной дисплазии (1:30 000-1:40 000); метафизарная хондро-дисплазия, тип Мак-Кьюсик (33 больных в 28 семьях); врожденная хлоридная диарея; врожденный нефротический синдром финского типа (1:8000); врожденная плоская роговица (52 больных); диастрофическая дисплазия (40 больных); синдром Ушера, тип III (157 больных); наследственная непереносимость фруктозы (16 больных); гиперорнитинемия со складчатой атрофией сосудистой оболочки и сетчатки (31 больной); инфантильный тип нейронального цероидного липофусциноза (1:13 000); лизинурическая непереносимость белка (1:60 000-1:80 000); синдром Меккеля (28 больных); карликовость Mulibrey (31 больной); синдром поражения мышц, глаз и мозга (14 больных); некетотическая гиперглицинемия (1:55 000); кистозная липомембранозная остеодисплазия со склеротической лейкоэнцефалопатией (13 больных); прогрессирующая миоклонус-эпилепсия (1:20 000); болезнь Салла (27 больных); избирательная мальабсорбция витамина В12 (27 больных). Кроме того, исследования, проведенные в Финляндии, показали очень низкие частоты в этой популяции больных ФКУ, муковисцидозом, цистинозом, гомоцистинурией, галактоземией, болезнями Тея-Сакса и Гоше.
Oсновной вывод о причинах накопления достаточно большого числа наследственных заболеваний в Финляндии по сравнению с другими популяциями сводится к особой популяционной истории финнов с момента заселения ими территории страны (Salmela E., 2012). Большинство предков современных финнов в течение первого тысячелетия нашей эры иммигрировали из Балтийского региона. Исходная популяция была небольшой. В течение последующих столетий население медленно увеличивалось и к XVII в. составило 400 000; в 1950 г. - 1,6 млн, а в 1970 г. - 4,6 млн человек. Таким образом, за три века население увеличилось в 11,5 раза. До недавнего времени население преимущественно было сельским, с низким уровнем миграционной активности, сохраняющимся до сих пор в сельской местности страны. Финны исходно образовали так называемый суперизолят на юге Финляндии. Резкий рост популяций при низком уровне обмена генами между ними способствовал проявлению эффективного дрейфа генов, эффекта родоначальника и последующего изменения генных частот в субпопуляциях, с накоплением некоторых редких генов и одновременной элиминацией других.
Предположение о том, что дрейф и «эффект родоначальника» явились причиной накопления наследственных болезней в Финляндии, удалось проверить при картировании генов этих болезней и обнаружении в них мутаций. По крайней мере для 16 аутосомно-рецессивных, 2 аутосомно-доминантных и Х-сцепленных заболеваний, частых в Финляндии, было продемонстрировано выраженное неравновесие по сцеплению между генами заболеваний и ДНК-маркерами, что можно рассматривать как подтверждение роли эффекта основателя в накоплении генов редких наследственных заболеваний в Финляндии (табл. 18-10).
При исследовании популяции евреев также было выявлено накопление ряда наследственных заболеваний (Goodman R.M., 1980).
Для понимания причин накопления наследственной патологии необходимо обратиться к историческим особенностям расселения евреев. Вследствие длительной географической разобщенности и традиционной национальной изоляции три большие группы евреев - ашкенази (82%), сефарды (11%) и восточные евреи (7%) - различаются по историческому происхождению, религии, языку. Все три группы отличаются между собой и по частотам и спектру наследственных болезней. К настоящему времени выявлено 120 наиболее характерных заболеваний у различных евреев, с преимущественно аутосомно-рецессивным типом наследования (в основном врожденные нарушения метаболизма).
Популяция восточных евреев, проживающих в арабских странах, на Кавказе и Индии, отличается высокой изолированностью и высокой частотой эндогамных браков. В этой популяции распространенными являются β-талассемия и недостаточность фермента глюкозо-6-фосфат-дегидрогеназы. Для евреев-сефардов, расселившихся в Северной Африке, Южной и Западной Европе и Америке, также характерно распространение с высокой частотой ряда наследственных болезней, однако отличных от тех, которые встречаются у ашкенази. Так, среди тунисских евреев найдено накопление избирательной мальабсорбции витамина В12 с протеинурией (q=0,02), среди марокканских - болезнь накопления гликогена, тип III (q=0,015) и атаксия-телеангиэктазия. Для евреев-сефардов характерна также высокая частота (от 1:600 до 1:5000) семейной средиземноморской лихорадки.
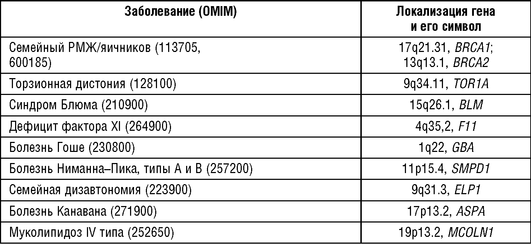
Среди ашкенази встречается целый ряд рецессивных заболеваний. Анализ аутосомно-рецессивных заболеваний выявил 9 нозологических форм, обнаруживающих высокую частоту в этой этнической группе евреев, в том числе болезнь Тея-Сакса (частота гетерозигот 3-4%), взрослая форма болезни Гоше (частота гетерозигот 5-6%), болезнь Канавана (частота гетерозигот 1,7-2%), болезнь Ниманна-Пика, типы А и Б (три мутации с частотой гетерозигот 1-2%), мукополи-липидоз (около 1%), синдром Блюма (1%), синдром Райли-Дея (3%), недостаточность фактора XI свертывания крови (две мутации - 8,1%), пентозурия (2,5-3%). Среди аутосомно-доминантных заболеваний - семейный РМЖ, обусловленный мутациями в генах BRCA1 и -2 (три мутации с общей популяционной частотой около 1%) и идиопатическая торзионная дистония (частота гетерозигот 0,1-0,3%).
У евреев-ашкенази основным механизмом накопления редкой наследственной патологии предполагается дрейф генов, для которого имелись исторические предпосылки, в частности диаспора, в ходе которой неоднократно возникали периоды резкого уменьшения численности евреев-ашкенази. Частоты так называемых «болезней ашкенази» варьируют в зависимости от географической подразделенности самой группы ашкенази. В то же время у евреев ашкенази, так же как у финнов, крайне редко встречается ФКУ и, по-видимому, другие наследственные болезни.
Накопление наследственных болезней в популяции евреев-ашкенази было использовано для картирования генов этих болезней, установления их молекулярной природы и выявления мутаций в этих генах, и в 2006 г. создана Национальная генетическая база данных Израиля, включающая фенотипические и генетические варианты у различных этнических групп страны. На 2016 г. в базе содержатся 2444 записи о наследственных болезнях среди израильских арабов, палестинских арабов и евреев (ашкенази, марокканских, иракских, иранских, йеменских, сефар-дов, евреев североафриканцев и евреев неизвестного происхождения) (Zlotogora J., 2017). База данных постоянно пополняется и корректируется. Последняя корректировка проведена в 2016 г. в соответствии с записями базы данных ClinVar (https:// www.ncbi.nlm.nih.gov/clinvar) и ExAC (http://exac.broadinstitute.org). Результаты молекулярно-генетического изучения некоторых наиболее частых наследственных болезней евреев-ашкенази представлены в табл. 18-11.
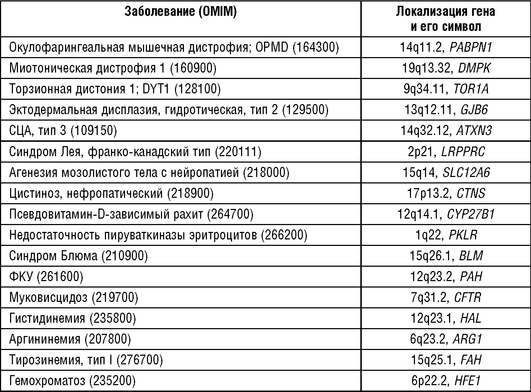
Дрейф генов и «эффект основателя» выступают в качестве основных причин накопления редкой наследственной как доминантной, так и рецессивной патологии в популяции франко-канадцев в Квебеке. Доказательство роли дрейфа генов и «эффекта основателя» получены для гидротической эктодермальной дисплазии, для тирозинемии I типа, псевдовитамин-D-зависимого рахита и ряда других наследственных заболеваний, обнаруживающих накопление у франко-канадцев. В табл. 18-12 представлены некоторые заболевания, обнаруженные c высокими частотами в популяции франко-канадцев (De Braekeeleer M., Dao T.-N., 1994 a, b).
Можно привести немало примеров накопления редких мутантных генов в популяциях, являющегося результатом действия особенностей генетической структуры популяций, в частности, генетического дрейфа: амавротическая идиотия юношеского типа, описанная Шегреном в шведском изоляте; атаксия Фридрейха, особый случай карликовости, обнаруженный Хангхартом в долинах Швейцарских Альп; синдром Вернера в Сардинии; фукозидоз в Италии; анемия Фанкони в ЮАР; алкаптонурия в Чехии; муковисцидоз в области Бретань во Франции и в Дании. В некоторых случаях на первый план в качестве причины дифференциации популяций по частотам наследственных болезней выступает «эффект родоначальника». Наиболее известный пример эффекта родоначальника - накопление двух аутосомно-доминантных заболеваний - семейной гиперхолестеринемии (частота гетерозигот 1%) и порфирии (0,3%) среди африканеров Южной Африки. Создана база данных NEMDB: National and Ethnic Mutation Database (Patrinos G.P., 2006), в которой содержится постоянно обновляемая обширная информация об описанной генетической гетерогенности группы или популяции, посвященная документированию существующей генетической гетерогенности в различных этнических группах населения в 57 разных странах.
Говоря о разнообразии наследственной патологии в больших этнических группах, нельзя ограничиваться только заболеваниями, обнаружившими локально высокие или локально низкие частоты в популяциях, как у финнов или евреев ашкенази. Эти болезни не исчерпывают собой всю наследственную патологию этих этнических групп и даже не составляют значительную часть спектра их наследственных болезней. Проведенное исследование в 1992 г. J. Verheij и L. ten Kate показало, что в Нидерландах, как следует из публикаций до 1992 г., зарегистрировано 672 различных менделевских фенотипа, из которых 321 фенотип приходился на аутосомно-доминантные, 283 - на аутосомно-рецессивные и 68 - на Х-сцепленные фенотипы. Полученные результаты легли в основу молекулярно-генетического изучения этих болезней, определены спектр и частоты мутаций основных заболеваний, характерных для данной популяции (проект «Геном Нидерландов» (GoNL) http://www.nlgenome.nl). Для четырех частых для Нидерландов аутосомно-рецессивных заболеваний [понтоцеребеллярная гипоплазия, тип 2 (PCH2), синдром Пена-Шокейра, тип I (FADS), ризомелическая точечная хондродисплазия, тип 1 (RCDP1), несовершенный остеогенез, тип IIB/III (OI)] проведен целевой (преконцепционный) скрининг брачующихся пар на носительство, показавший высокую эффективность (Mathijssen I.B., 2015).
Дифференциация по разнообразию наследственных болезней российских популяций
В России изучение дифференциации по разнообразию наследственных болезней в основном ограничивается анализом распространения отдельных заболеваний либо отдельных групп наследственных заболеваний. Например, в последние годы активно изучается распространенность наследственных заболеваний нервной системы в различных популяциях РФ: Амурской, Саратовской, Самарской, Владимирской и других областях. В Республике Башкортостан на основании изучения распространенности наследственных заболеваний нервной системы на базе медико-генетической консультации (МГК) созданы и функционируют регистры по нескольким заболеваниям: хорее Гентингтона, миотонической дистрофии, наследственной сенсомоторной нейропатии и т.д. Изучению наследственных заболеваний нервной системы способствует хорошие знания неврологов, высокий удельный вес и известная молекулярная природа большинства заболеваний данной группы. Oднако в отношении наследственных заболеваний других органов и систем (глаз, скелета, кожи, желез внутренней секреции) дело обстоит значительно хуже.
В 80-х годах прошлого столетия в Московском НИИ педиатрии и детской хирургии создан первый российский медико-генетический регистр «ГЕНРЕГ», функционировавший в нескольких учреждениях РФ. Данный регистр предназначался в основном для решения вопросов диспансеризации пораженных детей и обеспечения помощи в медико-генетическом консультировании семей. Oднако в отношении моногенной наследственной патологии аналитических статей по дифференциации в популяциях России наследственных заболеваний не было.
Созданы и функционируют регистры врожденной и наследственной патологии в Республике Якутия, в Томской области на базе МГК по результатам ежегодного приема и на основании экспедиционных данных. По результатам данных регистров показана высокая частота в якутской популяции как минимум трех заболеваний с аутосомно-доминантным типом наследования - спиноцеребеллярной атаксии 1-го типа (частота среди якутов 1:2590; ген SCA1), миотонической дистрофии (частота среди якутов 1:4695; ген DMPK), окулофарингеальной миопатии (частота среди якутов 1:11680; ген PABPN1). У якутов также выявлено накопление четырех заболеваний с аутосомно-рецессивным типом наследования - синдрома 3-М (частота среди якутов 1:8700; мутация G5741→A в гене NBAS), синдрома нанизма с субатрофией зрительных нервов, дистрофией сетчатки и пельгеровской аномалией лейкоцитов (частота среди якутов 1:9600; мутация R1914H в гене NBAS), мукополисахаридоз-плюс (частота среди якутов 1:4694; мутация c.1492C>T в гене VPS33A) и метгемо-глобинемии 1-го типа (частота среди якутов 1:5677; мутация c.806 C→T в экзоне 9 гена CYB5R3). Анализ гаплотипов для всех заболеваний показал, что причиной накопления является «эффект родоначальника» (Максимова Н.Р., 2008).
Сравнение разнообразия наследственных болезней одновременно с определением основных причин дифференциации российских популяций и этнических групп по генам по спектру наследственной патологии приведено в ряде работ лаборатории генетической эпидемиологии Федерального государственного бюджетного научного учреждения «Медико-генетический научный центр им. Академика Н.П. Бочкова». Проанализированы основные закономерности формирования спектра наследственных болезней в 11 этнических группах России: у русских (Кировской, Костромской, Брянской, Ростовской, Тверской областей, Краснодарского края), пяти народов Волго-Уральского региона (марийцев, чувашей, удмуртов, башкир и татар) и пяти народов Северного Кавказа (адыгейцев, черкесов, абазин, карачаевцев и ногайцев). Численность обследованного населения 13 регионов - около 3,7 млн человек. Сводный нозологический спектр наследственных болезней, выявленных в 13 российских регионах, составил 530 клинически различных заболеваний (242 аутосомно-доминантных, 226 аутосомно-рецессивных и 62 Х-сцепленных рецессивных форм) у 10 640 больных. По-видимому, число генетических нозологических форм в обследованных популяциях существенно больше, так как во многих случаях невозможно было точно дифференцировать клинические варианты генетически гетерогенных наследственных болезней, таких, например, как пигментный ретинит, врожденная катаракта, наследственная сенсорная нейропатия, несиндромальная нейросенсорная тугоухость и т.д. (Зинченко Р.А. и др., 2009, 2019).
Проанализировано накопление наследственных болезней в отдельных популяциях и/или этносах. Во всех популяциях диагностированы аутосомно-доминантные, аутосомно-рецессивные и Х-сцепленные заболевания, обнаружившие накопление или, наоборот, локально низкие значения распространенности отдельных наследственных болезней (Зинченко Р.А. и др., 2009, 2019).
В табл. 18-13 приводятся нозологический спектр и распространенность аутосомно-доминантных заболеваний (10-5 ), обнаруживших накопление (p <0,001) в обследованных популяциях России. Жирным шрифтом с подчеркиванием обозначена частота заболевания, отклоняющаяся от среднего значения. Заболевания расположены в соответствии с убыванием среднего значения.
Продолжение табл. 18.13
Продолжение табл. 18.13
Продолжение табл. 18.13
Окончание табл. 18.13
|
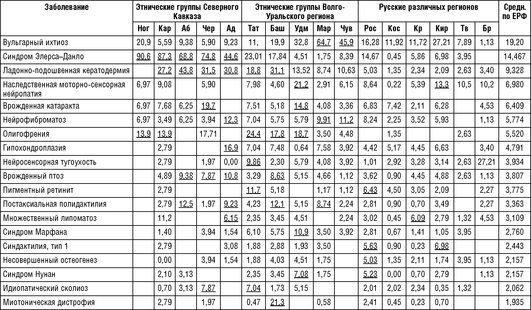
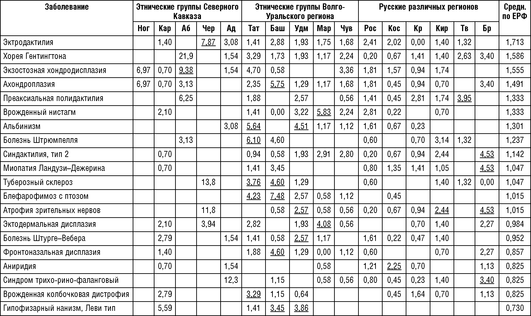
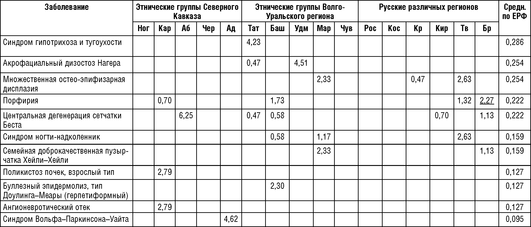
Примечание для табл. 18-13 - 18-15: Ног - ногайцы Карачаево-Черкесской Республики; Аб - абазины Карачаево-Черкесской Республики; Чер - черкесы Карачаево-Черкесской Республики; Кар - карачаевцы Карачаево-Черкесской Республики; Тат - татары Республики Татарстан; Баш - башкиры Республики Башкортостан; Удм - удмурты Республики Удмуртия; Map - марийцы Республики Марий Эл; Чув - чуваши Республики Чувашия; Ад - адыгейцы Республики Адыгея; Рос - Ростовская область; Кос - Костромская область; Крас - Краснодарский край; Кир - Кировская область; Tb -Тверская область; Бр - Брянская область; Средн. по ЕРФ - средние значения распространенности на всю рассматриваемую выборку Европейской части России.
Как следует из табл. 18-13, заболевания, обнаруживающие накопление в обследуемых популяциях, в большинстве случаев относятся к группе частых (встречающихся с распространенностью 1:50 000 и чаще) и условно частых (от 1:50 0001:100 000) заболеваний в российских популяциях. Однако немало случаев накопления отдельных нозологических форм, которые являются частыми только для данной этнической группы или конкретной популяции в рамках одного этноса.
Кроме того, анализ таблицы 18-13 выявил некоторые этнические особенности в распространении наследственных болезней. Например, ряд заболеваний у русских был диагностирован реже, чем в других этнических группах.
Ладонно-подошвенный гиперкератоз с более высокой распространенностью зарегистрирован среди карачаевцев, абазин, черкесов, адыгейцев, татар и башкир; вульгарный ихтиоз и брахидактилия типов Е и В - среди марийцев и чувашей; нейрофиброматоз 1-го типа чаще встречается среди марийцев, адыгейцев и чувашей; доминантная олигофрения - у башкир, татар, удмуртов, ногайцев и карачаевцев; врожденный птоз - среди абазин, черкесов, адыгейцев, башкир; синдром Элерса-Данло - среди 5 народов Северного Кавказа. Напротив, синдактилия типа 1, бородавчатый гиперкератоз, эссенциальный тремор, отосклероз и несовершенный остеогенез чаще встречались среди русского населения.
Для большинства клинических форм наследственных болезней в этнических популяциях определена генетическая гетерогенность (локусная), требующая для идентификации секвенирования клинического экзома или полногеномных исследований (наследственная моторно-сенсорная нейропатия, прогрессирующая мышечная поясно-конечностная дистрофия, пигментный ретинит, врожденная катаракта, младенческая эпилептическая энцефалопатия, метатропная дисплазия и т.д.). Выявлено большое количество ранее не описанных генетических вариантов с аутосомно-доминантным типом наследования.
К этим заболеваниям относятся летальный инфантильный остеопетроз (мутация c807+5G>A в гене TCIRG1), эритроцитоз (мутация с.С598Т в гене VHL) и наследственный врожденный гипотрихоз, марийский тип (мутация ΔΕχ 4 в гене LIPH). Проведенное популяционное изучение частот мутаций среди здоровых индивидов пяти этнических групп (русские, марийцы, чуваши, башкиры, удмурты) показало высокие значения мутации c807+5G>A в гене TCIRG1 (остеопетроз) только среди чувашского - 1,68% (расчетная частота заболевания 1:3900 новорожденных) и марийского населения - 0,84% (1:14 000) (Bliznetz E.A. et al., 2009). Мутация с. С598Т в гене VHL, характерная для развития эритроцитоза, выявлена в трех этнических группах: у чувашей частота мутации составила 1,84% (расчетная частота заболевания - 1:3000 новорожденных), для марийцев - 0,87% (расчетная частота - 1:13 150), для удмуртов - 0,47% (расчетная частота - 1:44 600).
Проведенное популяционное изучение частот мутаций среди здоровых индивидов пяти этнических групп (русские, марийцы, чуваши, башкиры, удмурты, татары) показало, что частота мутации ΔΕχ 4 гена LIPH (гипотрихоз) у чувашей достигает 2,72% (расчетная частота заболевания 1:1350 новорожденных), марийцев - 1,74% (1:2746), татар - 0,73% (1:18 832), удмуртов - 0,44% (1:51 020), башкир - 0,312% (1:103 092), среди русского населения мутаций не выявлено (Kazantseva A. et al., 2006). Дальнейшее молекулярно-генетическое исследование, проведенное на группе пациентов с клиническими проявлениями гипотрихоза, но не имеющих мутации ΔΕχ 4 в гене LIPH, методом NGS, позволило выявить гомозиготную, ранее не описанную мутацию c.712G>T (p.Val238Leu) в гене KRT25 и подтвердить ее секвенированием по Сэнгеру. Проведенный анализ на 300 образцах ДНК здоровых индивидов из трех популяций (чувашской, марийской, русской) установил, что частота генетического варианта c.712G>T (p.Val238Leu) в гене KRT25 для чувашской и марийской популяций составляет 1,5% (расчетная частота заболевания 1:4147) (Zernov N.V. et al., 2016).
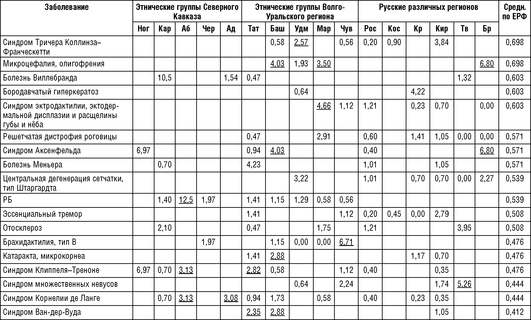
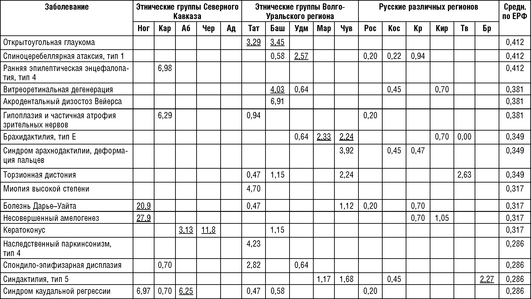
В табл. 18-14 представлены аутосомно-рецессивные заболевания, показавшие накопление в различных популяциях/этносах. Для аутосомно-рецессивных заболеваний также найдены некоторые этнические закономерности в их распространенности. Три аутосомно-рецессивных заболевания с высокой частотой выявлены в чувашской и марийской популяциях, практически не встречаясь в других регионах как РФ, так и мира. Для всех трех заболеваний картированы и идентифицированы гены и определены частоты мутаций генов в популяциях.
При подтверждающей молекулярно-генетической диагностике частых аутосомно-рецессивных заболеваний выявлена аллельная генетическая гетерогенность в разных этнических группах.
По данным неонатального скрининга, в Карачаево-Черкесской Республике определена очень высокая частота ФКУ/гиперфенилаланинемии, которая составила 1:850 новорожденных (только ФКУ 1:1581), средняя частота по России составляет 1:7000. Таким образом, выявлена самая высокая частота заболевания, зарегистрированная в мире. При проведении ДНК-исследования выявлены этнические особенности в частоте генетических вариантов в гене РАЯ. Наиболее частой мутацией в гене РАЯ для карачаевцев оказался генетический вариант R261*, аллельная частота которого среди представителей титульной нации - карачаевцы - составила 67,3%. Наиболее частая для европейских и русских популяций мутация R408W не выявлена ни у одного пациента с ФКУ/гипер-фенилаланинемией карачаевского происхождения. Определен спектр частых мутаций (R261*, R413P, V230I, P211T, F331S, P211L) для карачаевцев, позволяющий выявлять хотя бы одну мутацию у 95,7% больных. На основании проведенных исследований предполагается независимое происхождение мутации R261* на территории Карачаево-Черкесской Республики, так как выявлен общий гаплотип на хромосомах с мутацией R261* среди карачаевцев. Гаплотип гена PAH на хромосомах с мутацией R261* не совпадает с ранее описанными в литературе и подтверждает влияние «эффекта основателя» на накопление ФКУ в популяции Северного Кавказа (Gundorova P. et. al., 2018).
Особенности спектра и частот мутаций выявлены и при проведении молекулярно-генетической диагностики у пациентов с муковисцидозом ряда этнических групп Северного Кавказа. У карачаевцев из Карачаево-Черкессии определена высокая доля (92,86%) мутаций W1282X в гене CFTR, распространенная в РФ мутация F508del в гене CFTR не выявлена. Анализ частот гаплотипов полиморфизмов IVS1CA-IVS6aGATT-IVS8CA-IVS17bCA в выборке здоровых карачаевцев, не являющихся носителями мутации W1282X, показал, что наиболее частыми являются гаплотипы 22-7-16-13 (32%) и 23-7-16-13 (16%). Гаплотипы же, ассоциированные с мутацией W1282X у некарачаевцев, являются весьма редкими на нормальных хромосомах, составляя около 0,8% - для гаплотипа 26-7-17-17 и 1% - для гаплотипа 24-7-17-17, тогда как гаплотип 26-7-17-18, ассоциированный с мутацией W1282X у карачаевцев, на нормальных хромосомах исследуемой выборки не обнаружен. Можно предположить, что проникновение мутации W1282X в гене CFTR на территорию Карачаево-Черкессии и европейской части РФ происходило разными группами носителей этой мутации и, возможно, в разное время. На основании анализа гаплотипов можно предположить, что проникновение мутации W1282X на Кавказ связано с расселением евреев из Ирана в раннее средневековье, а ее высокая частота среди больных муковисцидозом у карачаевцев является следствием «эффекта основателя». Распространение мутации W1282X в восточно-европейских регионах России обусловлено расселением евреев ашкенази в более позднее время (Petrova N.V. et. al., 2016).
Окончание табл. 18.14
|
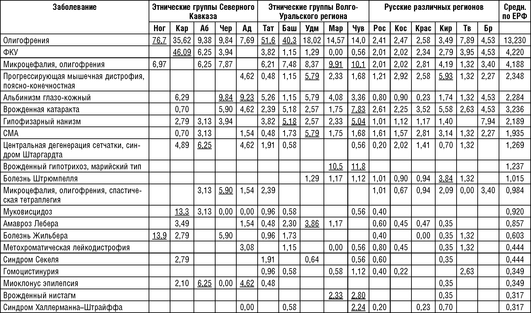
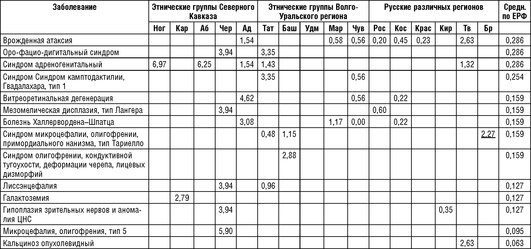
Особенный - уникальный - спектр мутаций обнаружен у чеченцев, больных муковисцидозом из Чеченской Республики. Определена высокая частота (более 90% мутантных аллелей в гене CFTR) двух мутаций: 1677delTA - 81,3% и E92K - 12,5%. Мутации 1677delTA и E92K оказались связаны с одним и тем же гаплотипом (22-7-1613) внутригенных ДНК-маркеров (IVS1CA, IVS6aGATT, IVS8CA и IVS17bCA). Этот гаплотип наиболее распространен среди нормальных хромосом здоровых чеченцев. Тот факт, что мутация 1677delTA была обнаружена у автохтонных этнических групп Кавказского региона (чеченцев, ингушей, грузин, армян) и связана с единым гаплотипом, может указывать на единый источник происхождения (или проникновения) этой мутации в Кавказский регион, а различия в частотах, наблюдаемые в различных популяциях, могут быть обусловлены дрейфом генов (Petrova N.V. et. al., 2019).
В табл. 18-15 представлены Х-сцепленные заболевания, показавшие накопление в различных этнических группах в регионах РФ. Как следует из табл. 18-15, дифференциация в частотах встречаемости зарегистрирована и для этой группы заболеваний.
С целью выяснения механизма, определяющего дифференциацию российских популяций по грузу наследственных болезней, а также формирующего очаги локально высоких частот отдельных нозологических форм, оценено влияние основных факторов микроэволюции (мутации, естественный отбор, миграции, ассортативность, дрейф генов) на медико-генетические характеристики изученных популяций.
В распространенности мутаций de novo, оцененной прямым методом, не выявлено различий по обследованным регионам (χ2 =1,68; D.f.=11; p >0,05). Распространенность варьировала в диапазоне от 0,13 (Костромская область) до 0,19 (Республика Башкортостан) на 1000. Средняя частота мутаций de novo на локус на поколение в обследованной выборке составила 0,495×10-5 .
Дифференциации между субпопуляциями не обнаружено и по такому фактору популяционной динамики, как естественный отбор, оцененный на основании анализа индекса Кроу, а также с помощью других популяционных статистик (Зинченко Р.А. и др., 2009).
Во всех регионах для оценки роли миграционных процессов, брачной ассортативности и дрейфа генов изучена брачно-миграционная структура (индекс эндогамии и локальный инбридинг через модель изоляции расстоянием Малеко) и оценены значения коэффициента инбридинга. В общей совокупности проанализировано более 90 000 брачных записей и тотально подвергнуты анализу фамилии (для расчета значений инбридинга) лиц репродуктивного периода (старше 18 лет). В обследованных регионах наблюдалась отрицательная брачная ассортативность, и, следовательно, причиной генетической дифференциации популяций по частотам генов не мог служить неслучайный инбридинг. Значения случайного инбридинга (F) оказались низкими по абсолютным значениям, но варьировали в очень широких пределах, от практически 0 в субпопуляциях Краснодарского края до 28,12×10-3 в Теудчежском районе Республики Адыгея. Средневзвешенное значение случайного инбридинга для городских популяций составило 0,57×10-3 , а для сельских - 4,36×10-3 . В то же время как в городских, так и в сельских популяциях выявлена выраженная изменчивость значений случайного инбридинга между отдельными популяциями. Значения индекса эндогамии, отражающего как миграционную активность населения, так и степень изолированности популяций, также значительно различались - от 0 в г. Козьмодемьянске Республики Марий Эл до 0,97 в Балтачевском районе Республики Башкортостан. Сельские популяции в большинстве случаев представляют современные, в определенной мере изолированные популяции с индексом эндогамии от 0,55 до 0,97. Столь высокие значения индекса эндогамии, несомненно, являются предпосылкой для накопления инбридинга (эффективная численность этих популяций варьировала от 3,5 до 30 000 человек).
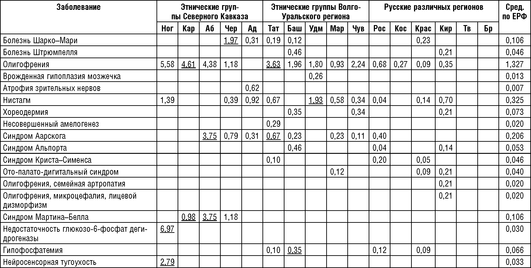
На основе изученных корреляций, проведенных в разных группах (1-я группа - между грузом аутосомных заболеваний и случайным инбридингом; 2-я группа - между грузом и индексом эндогамии; 3-я группа - между коэффициентами накопления аутосомных заболеваний и случайным инбридингом; 4-я группа - между коэффициентами накопления аутосомных заболеваний и индексом эндогамии), было показано, что основной причиной генетической дифференциации популяций по грузу наследственных болезней являются подразделенность популяций, связанный с ней дрейф генов и «эффект родоначальника».
Из всего вышесказанного можно заключить, что выявленные различия между отдельными популяциями и этническими группами в распространенности отдельных наследственных заболеваний тесно связаны с особенностями этногенеза и истории формирования популяций, а гены наследственных болезней могут служить мощным инструментом для характеристики этногенетических процессов, происходящих в популяциях.
Список литературы
-
Адрес в Интернете сайта «Online Mendelian Inheritance in Man» http://www.ncbi. nlm.nih.gov/Omim/
-
Адрес в Интернете сайта «urpha.net» http://www.orpha.net/
-
Адрес в интернете «Финская база данных по болезням» http://www.findis.org/
-
Алтухов Ю.П. Генетические процессы в популяциях. М.: Наука, 2003. 370 с.
-
Гинтер Е.К., Зинченко Р.А. и др. Генетическая структура и наследственные болезни Чувашской популяции. Коллективная монография / Под ред. Е.К. Гинтера, Р.А. Зинченко. Чебоксары: Пегас, 2006. 232 с.
-
Гинтер Е.К. Медицинская генетика. М.: Медицина, 2003. 447 с.
-
Животовский Л.А. Популяционная биометрия. М.: Наука, 1991. С. 128-130.
-
Зинченко Р.А., Ельчинова Г.И., Гинтер Е.К. Факторы, определяющие распространение наследственных болезней в российских популяциях // Медицинская генетика. 2009. Т. 8, № 12(90). С. 7-23.
-
Зинченко Р.А., Куцев С.И., Александрова О.Ю., Гинтер Е.К. Основные методологические подходы к выявлению и диагностике моногенных наследственных заболеваний и проблемы в организации медицинской помощи и единых профилактических программ // Проблемы социальной гигиены, здравоохранения и истории медицины. 2019. Т. 27, № 5. С. 865-877. doi: http://dx.doi.org/10.32687/0869-866X-2019-27-5-865-877
-
Максимова Н.Р., Сухомясова А.Л., Гуринова Е.Е. и др. Генетико-эпидемиологические и социально-экономические аспекты наследственной этноспеци-фической патологии в Якутии // Медицинская генетика. 2008. Т. 7, № 10(76). С. 35-43.
-
Мерфи Э.А., Чейз Г.А. Основы медико-генетического консультирования. М.: Медицина, 1979. 215 с.
-
Ратнер В.А. Математическая популяционная генетика. Новосибирск: Наука, 1977. 55 с.
-
Тимофеев-Ресовский Н.В., Яблоков А.В., Глотов Н.В. Очерк учения о популяциях. М.: Наука, 1973. 277 с.
-
Baird P.A., Anderson T.W., Newcombe H.B., Lowry R.B. Genetic disorders in children and young adults: a population study // Am. J. Hum. Genet. 1988. Vol. 42. Р. 677-693.
-
Bliznetz E.A., Tverskaya S.M., Zinchenko R.A. et al. Genetic analysis of autosomal recessive osteopetrosis in Chuvashiya: the unique splice site mutation in TCIRG1 gene spread by the founder effect // European Journal of Human Genetics. 2009. Vol. 17. P. 664-672. doi:10.1038/ejhg.2008.234
-
Carter C.O. Monogenic disorders // J. Med. Genet. 1977. Vol. 14. P. 316-320.
-
De Braekeeleer M., Dao T.-N. Hereditary disorders in French Canadian population of Quebec. In search of founders // Hum. Biol. 1994. Vol. 66. P. 205-224.
-
De Braekeeleer M., Dao T.-N. Hereditary disorders in French Canadian population of Quebec. II. Contributions of Perche // Hum. Biol. 1994. Vol. 66. P. 225-250.
-
Edwards A., Cavalli-Sforza L.L. Affinity as revealed by differences in gene frequencies / In: The Assessment of Population Affinities in Man / Ed. by: J.S. Weiner, I. Huisinda. Oxford: Charendon Press, 1972. P. 37-47.
-
Goodman R.M. Genetic Disorders among the Jewish People. Baltimore: The John Hopkins Univ. Press, 1980. P. 965-970.
-
Crowgey E.L., Washburn M.C., Kolb E.A., Puffenberger E.G. Development of a Novel Next-Generation Sequencing Assay for Carrier Screening in Old Order Amish and Mennonite Populations of Pennsylvania // J Mol Diagnostics. 2019. Vol. 21, N 4. Р. 687-694. doi: 10.1016/j.jmoldx.2019.03.004.
-
Gundorova P., Zinchenko R.A., Kuznetsova I.A. et al. Molecular-genetic causes for the high frequency of phenylketonuria in the population from the North Caucasus // PLoS ONE. 2018. Vol. 13, N 8. P. e0201489. doi: 10.1371/journal.pone.0201489
-
Jones A., Bodmer W.F. Our future inheritance: choice or chance? A study by a British Association Working Party. London: Oxford Univ. Press, 1974. 141 p.
-
Kazantseva A., Goltsov A., Zinchenko R. et al. Human Hair Growth Deficiency Is Linked to a Genetic Defect in the Phospholipase Gene LIPH // Science. 2006. Vol. 314, N 5801. P. 982-985. doi: 10.1126/science.1133276
-
Malecot G. Isolation by distance. Genetic Structure of Population / Ed. N.E. Morton. Honolulu: Univ. of Hawaii Press, 1973. P. 72-75.
-
Mathijssen I.B., Henneman L., van Eeten-Nijman J.M. et al. Targeted carrier screening for four recessive disorders: High detection rate within a founder population // European Journal of Medical Genetics. 2015. Vol. 58, N 3. P. 123-128.
-
Matsunaga E. Incidence of Genetic disease in Japan // Human Genetics. Berlin, etc.: Springer-Verlag, 1986. 716 p.
-
McKusick V.A. Ethnic distribution of disease in non-Jews // Isr. J. Med. Sci. 1973. Vol. 9. P. 1375-1382.
-
Muller H.J. Our load of mutation // Am. J. Hum. Genet. 1950. Vol. 2. P. 111-176.
-
Neel J.V. Mutation and disease in man // Can. J. Genet. Cytol. 1978. Vol. 20. P. 295306.
-
Norio R., Nevanlinna H.R., Perheentupa J. Hereditary diseases in Finland: rare flora in rare soul // Ann. Clin. Res. 1973. Vol. 5. P. 109-141.
-
Patrinos G.P. National and ethnic mutation databases: recording populations' genog-raphy // Hum Mutat. 2006. Vol. 27, N 9. P. 879-887.
-
Peltonen L. Molecular background of the Finnish disease heritage // Ann. Med. 1997. Vol. 29. P. 553-556.
-
Petrova N.V., Kashirskaya N.Yu., Vasilieva T.A. et al. High proportion of W1282X mutation in CF patients from Karachai-Cherkessia // Journal of Cystic Fibrosis. 2016. Vol. 15, N 3. P.e28-e32. DOI: 10.1016/j.jcf.2016.02.003.
-
Petrova N.V., Marakhonov A.V., Vasilyeva T.A. et al. Comprehensive genotyping reveals novel CFTR variants in cystic fibrosis patients from the Russian Federation// Clinical Genetics. 2019. Vol. 95, N 3. Р. 444-447. doi: 10.1111/cge.13477
-
Picheral H. La mobilite geographique des francais du XVI an XIX siecle // Genet. Pop. Hum.: Colog. INSERM, Toulouse, 1985. Vol. D, N. 8. P. 611-613.
-
Ratha-Rama D.A., Rao N. Inbreeding and the incidence of childhood genetic disorders in Karnataka, South India // J. Med. Genet. 1987. Vol. 24. P. 362-365.
-
Ruiz-Perez V.L., Ide S.E., Strom T.M. et al. Mutations in a new gene in Ellis-van-Creveld syndrome and Weyers acrodental dysostosis // Nat. Genet. 2000. Vol. 6, N 24. P. 283-286.
-
Salmela E. Genetic structure in Finland and Sweden: Aspects of Population History and Gene Mapping // Helsinki: ed. Unigrafia, 2012. 136 р.
-
Sankaranarayanan K. Ionizing radiation and genetic risks IX. Estimates of the frequencies of Mendelian diseases and spontaneous mutation rates in human populations: a 1998 perspective // Mutat. Res. 1998. Vol. 411. P. 129-178. doi: 10.1016/S1383-5742(98)00012-X
-
Stevenson A.C. The load of hereditary defects in human populations. // Rad. Res. 1959. Vol. 1. P. 306-325.
-
Teebi A.S., Farad T.I. Genetic disorders among Arab population // Oxford Monographs on Medical Genetics. Oxford: Oxford Univ. Press, 1997. Vol. 30. Р. 36-52.
-
Trimble B.R., Smith M.E. The incidence of genetic disease and the impact on man of an altered mutation rate // Can.J. Genet.Cytol. 1977. Vol. 19. P. 375-385.
-
UNSCEAR: Sources and effects of ionizing radiation. United Nations Scientific Committee on the Effects of Atomic Radiation, 1977. Report to the General Assembly, United Nations, New York.
-
UNSCEAR: Genetic and somatic effects of ionizing radiation. United Nations Scientific Committee on the Effects of Atomic Radiation, 1986. Report to the General Assembly with annexes, United Nations, New York.
-
Wright S. The origin of the F-statistics for describing the genetic aspects of population structure. Genetic structure of populations / Ed. Morton E.D. Honolulu: University of Hawaii Press, 1973. P. 3-36.
-
Zernov N.V., Skoblov M.Y., Marakhonov A.V. et al. Autosomal Recessive Hypotrichosis with Woolly Hair Caused by а Mutation in the Keratin 25 Gene Expressed in Hair Follicles // J. Invest. Dermatol. 2016. Vol. 136, N 6. P. 1097-105.
-
Zlotogora J., Patrinos G.P. The Israeli National Genetic Database: a 10-year experience // Hum Genomics. 2017. Vol. 11. Р. 5. doi: 10.1186/s40246-017-0100-z.