Медицинская генетика : национальное руководство / под ред. Е. К. Гинтера, В. П. Пузырева, С. И. Куцева. - Москва : ГЭОТАР-Медиа, 2022. - 896 с. (Серия "Национальные руководства") - ISBN 978-5-9704-6307-9 |
Аннотация
Настоящее издание подготовлено коллективом авторов, абсолютное большинство которых хорошо известны не только отечественному, но и зарубежному читателю.
В книге представлены основные разделы генетики человека и медицинской генетики, такие как генетика развития человека, эпигенетика, генетика соматических клеток, популяционная генетика, фармакогенетика, иммуногенетика и система генов HLA. Также описаны современные представления об основных классах наследственных болезней: моногенных, включая популяционную генетику моногенных заболеваний, наследственных болезнях обмена веществ, митохондриальных заболеваниях, патологиях, обусловленных расширением зоны тандемных тринуклеотидных повторов, хромосомных болезнях, включая те, что обусловлены микрохромосомными перестройками, заболеваниях, связанных с геномным импринтингом, многофакторных болезнях, врожденных пороках развития и тератогенных синдромах.
Отдельная часть руководства посвящена лечению и профилактике наследственных болезней, а также этическим проблемам медицинской генетики. В ней подробно рассмотрена проблема генной терапии, в том числе с использованием методов редактирования генома, которые многими исследователями рассматриваются как наиболее перспективные в генотерапии заболеваний человека.
Книга будет полезна врачам практически всех специальностей с учетом все возрастающего вклада генетики в современную медицину, без которого невозможно ее дальнейшее совершенствование.
Глава 17. Моногенные наследственные болезни
Введение
Наиболее важным направлением исследований в медицинской генетике является выяснение генетической основы наследственных болезней и болезней с наследственным предрасположением. Практическим следствием таких исследований является разработка методов профилактики и лечения заболеваний. В этой главе мы изложим феноменологию проявления генов моногенных наследственных заболеваний, т.е. таких заболеваний, этиология которых обусловлена мутациями в отдельных генах. Моногенные наследственные болезни занимают особое место в патологии человека в силу лучшей изученности их природы по сравнению с другими типами наследственной патологии - хромосомной или мульти-факторной. Большинство из них встречается редко или даже очень редко, но суммарно, как следует из наших данных, основывающихся на широких генетико-эпидемиологических исследованиях, проведенных во многих популяциях европейской части России, распространенность моногенных наследственных болезней в российских популяциях может достигать 1% и более.
установление типа наследования моногенных заболеваний
Прежде, чем мы перейдем к изложению феноменологии проявления генов, вызывающих моногенные, или менделирующие, наследственные заболевания кратко остановимся на установлении типа их наследования, так как именно это является доказательством характера генетической природы таких заболеваний.
Основными понятиями при рассмотрении менделевского наследования признаков и болезней у человека являются «фенотип» и «генотип». Под термином фенотип принято понимать сумму всех внешних характеристик человека. Следует отметить, что когда говорится о внешних характеристиках, то следует подразумевать не только такие внешние признаки, как рост, цвет глаз или число пальцев на руках и ногах, но и различные физиологические, биохимические и даже молекулярные характеристики, которые могут измениться в результате действия генов. Фенотипические признаки, с которыми сталкивается медицинская генетика, - это наследственные болезни и их симптомы.
Под термином генотип (геном) понимают сумму всех генов организма. Однако нередко понятие «генотип» используют для описания состояния отдельного или нескольких генов. Генотип в значительной мере определяет фенотип, а гены - отдельные фенотипические признаки.
Фенотип, в отличие от генотипа, может меняться в течение жизни, генотип при этом остается постоянным, свидетельство этому - наш собственный онтогенез. В течение жизни внешне человек меняется, старея, а генотип - нет. За одним и тем же фенотипом могут стоять разные генотипы, и, напротив, при одном и том же генотипе фенотипы могут различаться. Последнее утверждение подкреплено изучением МБ. Их генотипы идентичны, а фенотипически они могут различаться по массе тела, росту, поведению и т.д. Из этого следует, что не существует в абсолютном большинстве случаев однозначной связи между генами и фенотипическими признаками, и проявление генов может изменяться как под действием факторов окружающей среды, так и в результате взаимодействия с другими генами. Очевидно, что между симптомами наследственного заболевания, такими как отсутствие ушной раковины, судороги, умственная отсталость, кисты в почках и многими другими, и изменением одного белка в результате мутации в каком-то конкретном гене - дистанция огромного размера. Мутантный белок - продукт мутантного гена - должен каким-то образом взаимодействовать с сотнями, если не с тысячами других белков, кодируемых другими генами, чтобы в результате изменился какой-то нормальный признак или появился патологический. Кроме того, продукты генов, принимающих участие в становлении любого фенотипического признака, могут взаимодействовать и модифицироваться факторами окружающей среды. Вместе с тем в случае, когда речь идет о моногенно наследуемых признаках (например, наследственных болезнях), действие мутантного гена не затушевывается многочисленными взаимодействиями его продукта с продуктами других генов или с факторами окружающей среды, и наличие мутантного гена(-ов) определяет изменение фенотипа. Это происходит, как в сложном механизме, когда дефекта одной детали достаточно, чтобы при тысячах других нормальных деталях механизм не работал.
Моногенное наследование признаков у человека называют еще менделевским, так как правила, согласно которым оно происходит, установил Грегор Мендель еще в 1865 г.
Особенности проявления менделевских правил наследования у человека
У человека, по понятным причинам, невозможны контролируемые скрещивания. Кроме того, число детей в семье, как правило, бывает небольшим. Для того чтобы доказать аутосомно-доминантный или аутосомно-рецессивный характер наследования заболевания, необходимо собрать достаточно большое число семей с большим числом детей в них. Если учесть, что абсолютное большинство наследственных заболеваний встречается редко, то становится ясно, что чаще всего медицинскому генетику приходится пользоваться упрощенными требованиями к доказательствам определенного типа наследования. Эти упрощенные требования следуют из менделевских правил наследования. Правомерно будет сказать, что заключение при таком типе анализа сводится к тому, что в исследуемой родословной сегрегация того или иного заболевания не противоречит определенному менделевскому типу наследования.
Обычно в поле зрения медицинского генетика попадает семья, в которой есть больной или больные с предположительно наследственным заболеванием. Для такой семьи обычно собирается родословная, т.е. устанавливаются предки родителей и их родственники и описывается статус их здоровья, а также другие сведения. Принято представлять родословную графически, используя для этого определенные символы, изображенные на рис. 17-1.
В 1991 г. Национальным обществом генетиков-консультантов (США) была создана специальная группа для стандартизации символов родословной. Эта группа подготовила рекомендации, которые после тщательного рецензирования были приняты как стандарт и опубликованы в двух журналах: American Journal Human Genetics и Journal of Genetic Counseling в 1995 г. С тех пор стандарты символов родословной получили мировое признание. В 2008 г. Bennet и соавт. проанализировали, как применяется стандартная номенклатура составления родословных 1995 г., используя для этого разные источники - литературу (книги и журналы), интервью с представителями обществ медицинских и клинических генетиков разных стран, и пришли к выводу, что эта номенклатура получила повсеместное распространение и практически не требует коррекции.
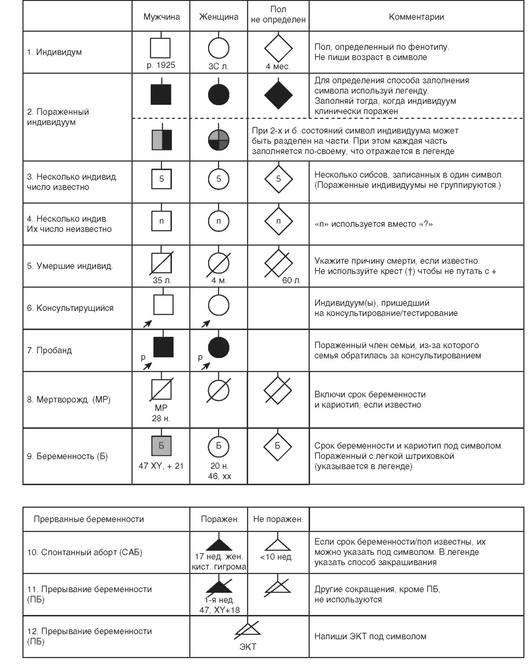
«Пробанд» - это термин, который обозначает пораженного члена семьи (живого или умершего) или плод при пренатальном медико-генетическом консультировании, из-за которого возникла необходимость обращения к врачу-генетику. В специально разработанную генетическую карту, которая является аналогом обычной истории болезни, подробно записываются сведения о больном. В дальнейшем продолжают сбор данных о родственниках первой степени родства (дети, сибсы и родители); затем второй степени родства (полусибсы, тети и дяди, племянники и племянницы, деды, бабушки и внуки); затем третьей степени родства (двоюродные сибсы). Врач должен собрать не только сведения, касающиеся конкретного патологического признака в семье, но и информацию о других серьезных заболеваниях и аномалиях, встречающихся среди членов семьи. Важно получить сведения о спонтанных абортах, мертворождениях и ранней детской смертности. Иногда они могут иметь прямое отношение к существу вопроса и сыграть важную роль при оценке как диагноза заболевания, так и прогноза, а также оценке риска. Например, множественные спонтанные аборты, особенно на раннем сроке беременности, позволяют предположить, что один из родителей является носителем сбалансированной хромосомной перестройки. Ранняя детская смертность, особенно сопровождающаяся рвотой, желтухой, судорогами и др., позволяет заподозрить наследственные болезни обмена веществ. Всегда нужно фиксировать основные медико-генетические данные с обеих сторон (по отцовской и материнской линии), даже если речь идет об аутосомно-доминантном заболевании, унаследованном от одного из родителей. На основании собранных сведений строят графическую схему родословной, используя специальные символы. Для этого могут быть применены заранее подготовленные шаблоны либо специальные пластиковые линейки для рисования, в которых есть кружки, квадраты, треугольники, стрелки разных размеров. Родословную сначала лучше рисовать карандашом, что позволяет вносить в нее изменения, о которых консультирующийся вспоминает позже. Затем такая родословная может быть обведена чернилами. В настоящее время существуют компьютерные программы для составления родословных.
Именно такая родословная становится предметом анализа медицинского генетика.
В 1966 г. появилось первое издание книги В. Мак-Кьюсика «Менделевское наследование у человека. Каталог аутосомно-доминантных, аутосомно-рецессивных и Х-сцепленных фенотипов», в котором были собраны сведения о различных, нормальных и патологических, состояниях у человека, которые обнаруживают менделевское наследование. В абсолютном большинстве случаев установление типа наследования заболеваний, представленных в этом каталоге, основывалось на указанных выше принципах. В связи с нестрогостью этих принципов некоторые заболевания переходили из одного каталога в другой по мере накопления материала и появления новых изданий. В настоящее время в этой книге, которая перестала переиздаваться с 1994 г. и существует в постоянно пополняющейся версии в Интернете (адрес сайта: http://www.ncbi.nlm.nih.gov/Omim/), представлены описание и библиография более 25 000 фенотипов, из них с установленным типом наследования и известной молекулярной основой фенотипа - около 6000.
Основную часть описанных фенотипов составляют те, что наследуются аутосомно-доминантно и аутосомно-рецессивно, их более 5400. Стоит добавить, что заболевания с этими типами наследования составляют в соответствующих каталогах примерно половину списков. За почти 54 года, прошедших с момента первого издания книги Мак-Кьюсика, число описаний менделирующих фенотипов у человека выросло более чем в 10 раз.
Аутосомно-доминантное наследование
В настоящее время известно несколько тысяч аутосомно-доминантно наследующихся фенотипов. Мы остановимся на некоторых общих принципах их выявления с помощью генетического подхода.
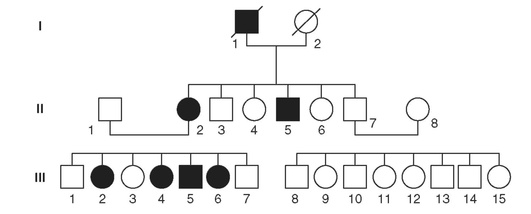
На рис. 17-2 представлена родословная, в которой наследуется синдром Марфана - аутосомно-доминантное заболевание, ген которого FBN1 (ген фибриллина) картирован в хромосоме 15. Синдром Марфана проявляется системным поражением соединительной ткани, так как дефектный белок входит в состав межклеточного вещества соединительной ткани. Больные с синдромом Марфана обычно высокого роста, с длинными конечностями и арахнодактилией пальцев. Кроме того, у больных наблюдаются кифосколиоз, миопия высокой степени или подвывих хрусталика, расширение корня аорты.
В родословной три поколения. В первом поколении был болен отец (I1), но к моменту исследования семьи он умер. Среди семи его детей двое больных (дочь - II2 и сын - II5) и пятеро здоровых. II2 состоит в браке со здоровым мужчиной, у них семеро детей, из которых больны четверо (III2, III4, III5, III6) - три девочки и один мальчик, а трое здоровы. Здоровый сын первого больного (II7) состоит в браке со здоровой женщиной (II8). У них восемь детей, все они здоровы.
Приведенная выше родословная демонстрирует требования, предъявляемые к родословным с аутосомно-доминантным наследованием. Во-первых, один из родителей больных детей также должен быть болен. Во-вторых, болезнь должна встречаться у людей обоего пола. В-третьих, согласно менделевским правилам наследования, половина детей больного родителя должна быть больна, и риск, который составляет 50%, остается постоянным для каждого последующего ребенка. Однако данное требование нежесткое, поскольку при небольшом числе детей в семье могут случайным образом наблюдаться заметные отклонения от ожидаемого отношения больных детей к здоровым. В-четвертых, должна наблюдаться передача заболевания от отца к сыну, что исключает сцепленное с полом наследование. В-пятых, у здоровых потомков больного все дети должны быть здоровы.
В действительности даже в тех случаях, когда сталкиваются с аутосомно-доминантным заболеванием в семье, далеко не всегда удается наблюдать родословную, подобную той, что приведена на рис. 17-2. Для этого есть несколько причин.
Одна из основных - это вновь возникающие мутации в отдельных ПК одного из родителей (при выполнении программы «Геном человека» установлено, что в большинстве случаев новые мутации возникают в гаметогенезе у мужчин). При таком наследовании больной с аутосомно-доминантной патологией будет единственным случаем заболевания в семье. Очевидно, что для такого больного шанс передать мутацию своим детям будет обычным для аутосомно-доминантного типа наследования, т.е. равен 50%.
Вторая причина отклонения от правил аутосомно-доминантного наследования - мозаицизм зародышевых клеток (мозаицизм означает присутствие двух или более генетически различных линий клеток в одном организме). Такой мозаицизм возникает на ранних стадиях развития организма в результате появления мутации в одной из клеток зародышевого пути в момент его обособления. Поскольку клетки зародышевого пути клонируются, мутация может оказаться в большей или меньшей части зрелых ПК, и как результат возможно появление в семье здоровых родителей детей с аутосомно-доминантными заболеваниями. Именно поэтому соматическим мозаицизмом объясняются повторные случаи ахондроплазии, несовершенного остеогенеза и других аутосомно-доминантных заболеваний у детей, родители которых клинически совершенно здоровы.
Аутосомно-рецессивное наследование
По данным «Каталога наследственных признаков человека» (OMIM), известно несколько тысяч фенотипов с аутосомно-рецессивным типом наследования. Примерно 50% этих фенотипов представлено болезнями, наследующимися аутосомно-рецессивно. Как и аутосомно-доминантные, аутосомно-рецессивные заболевания поражают все органы и системы человека и, следовательно, чрезвычайно разнообразны по своим проявлениям.
В силу редкости аутосомно-рецессивных заболеваний, а также тяжести течения многих из них в абсолютном большинстве случаев родители больных детей являются гетерозиготными носителями и клинически здоровы. 1/4 детей в браке гетерозиготных родителей должны быть гомозиготами по нормальному доминантному аллелю, 1/2 - гетерозиготами и 1/4 - гомозиготами по мутантному аллелю. Риск появления больного с аутосомно-рецессивным заболеванием в семье, где родители являются гетерозиготными носителями мутантного гена, составляет 25% и не меняется для любой беременности этой супружеской пары. Родословные больных с аутосомно-рецессивным заболеванием обычно невыразительны: родители и все ближайшие родственники больного здоровы, могут быть больны братья и сестры (оба пола поражаются одинаково часто). Если больной с рецессивным заболеванием вступает в брак, то его партнер в большинстве случаев является нормальной гомозиготой, и поэтому все дети в таком браке здоровы, но являются гетерозиготными носителями. Нередко в родословных с аутосомно-рецессивными заболеваниями родители больных оказываются близкими родственниками (рис. 17-3).
На рис. 17-3 представлена родословная, в которой наследуется редкое аутосомно-рецессивное заболевание - синдром Коккейна (OMIM 216400). Характерными признаками синдрома Коккейна являются карликовость, раннее старение, пигментная дегенерация сетчатки, атрофия зрительных нервов, тугоухость, умственная отсталость, повышенная чувствительность к свету и др. Ген синдрома картирован на хромосоме 5. Его белок участвует в репарации тиминовых димеров ДНК, возникающих после воздействия ультрафиолета.
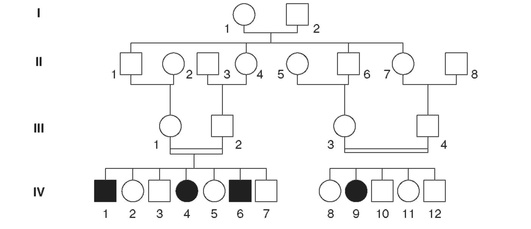
Родители больных детей, как и III2, так и III3 и III4, являются двоюродными сибсами. Кроме того, обе семьи с больными детьми родственны друг другу, так как II1, II4, II5 и II8 являются сибсами. Объяснение большей частоты близкородственных браков в семьях, где есть больные с аутосомно-рецессивным заболеванием, очень простое. Чем реже рецессивный ген встречается в популяции, тем меньше шансов, что оба родителя будут гетерозиготами по этому гену, так как вероятность такой супружеской пары равна произведению вероятностей возможности быть гетерозиготным носителем для каждого партнера. Так, при частоте гетерозиготного носительства гена ФКУ, равной примерно 0,02, вероятность брака гетерозиготных носителей составит 0,0004. Иными словами, одна из 2500 супружеских пар представлена гетерозиготными носителями (так как риск рождения больного ребенка для такой пары составляет 1/4, частота ФКУ в популяции как раз и составляет 1:10 000). В том случае, когда один из супругов является гетерозиготным носителем гена ФКУ (вероятность 0,02), а второй супруг является ему родственником, вероятность для второго супруга быть гетерозиготным носителем того же гена зависит от степени родства супругов. Для двоюродных сибсов она составляет, например, 25%, что в 12,5 раза выше, чем если бы супруги не были родственниками. Из приведенных расчетов также следует, что чем реже встречается рецессивный ген в популяции, тем большую долю среди семей с больными детьми будут составлять семьи, родители в которых являются близкими родственниками.
Итак, для аутосомно-рецессивно наследуемого признака характерно: носитель аутосомно-рецессивного признака является гомозиготой по мутантному аллелю соответствующего гена (если мутантные аллели у такого носителя являются разными, то его называют компаундной гетерозиготой), родители индивидуума с аутосомно-рецессивно наследуемым признаком (заболеванием) этого признака не имеют, но являются носителями мутантного гена. Признак могут проявить только братья и сестры (сибсы) этого индивидуума. Доля сибсов с рецессивным признаком в такой семье составляет 25%. Рецессивно наследуемый признак проявляется одинаково у сибсов разного пола. Для редких аутосомно-рецессивных заболеваний характерно, что родители больных детей значительно чаще, чем если бы это происходило случайно, являются близкими родственниками.
Наследование, сцепленное с Х-хромосомой
Гены, локализованные в Х-хромосоме, как и признаки, которые они контролируют, называются Х-сцепленными. Число известных Х-сцепленных заболеваний меньше, чем аутосомных, гены которых разбросаны по 22 аутосомам. Тем не менее известно около 300 генов, локализованных в Х-хромосоме, мутации в которых вызывают наследственные болезни. Всего же в Х-хромосоме выявлено более 600 генов, и, вероятно, это число не окончательное.
Прежде чем перейти к изложению наследования, сцепленного с Х-хромосомой, и особенностям родословных при этих типах наследования, следует напомнить, что у человека пол гетерогаметен. Женщины имеют во всех клетках две Х-хромосомы, а мужчины - одну Х- и одну Y-хромосому. Y-хромосома не гомологична Х-хромосоме, в ней содержится небольшое число генов. Мужчины являются гемизиготами по Х-хромосоме и большинству содержащихся в ней генов. Наследование половых хромосом происходит так же, как и простых менделевских признаков.
При случайном объединении гамет должно образоваться равное количество зигот женского и мужского пола.
Поведение генов, которые расположены в половых хромосомах, строго соответствует поведению самих хромосом. Если мутантный ген находится в одной из Х-хромосом матери (например, в хромосоме Х1), то эту Х-хромосому получат половина сыновей и половина дочерей матери-носительницы. Если ген отвечает за рецессивное заболевание, то сама мать должна быть здорова, так как у нее есть вторая нормальная Х-хромосома, но заболевание разовьется у той половины сыновей, которые получили хромосому Х1 с мутантным геном, поскольку Y-хромосома негомологична Х-хромосоме, и в ней просто не содержится пар абсолютному большинству генов Х-хромосомы. У дочерей, получивших от матери измененную хромосому, заболевание также не разовьется, так как они получат нормальную вторую Х-хромосому от отца. Таким образом, если мать является носительницей сцепленного с Х-хромосомой рецессивного гена, риск быть больным у ее сыновей составит 1/2, или 50%, а у ее дочерей - 0%. В то же время риск быть гетерозиготными носительницами у дочерей равен 50%.
Для рецессивных Х-сцепленных заболеваний характерно, что наследуют их мужчины - родственники друг другу по материнской линии. Это могут быть двоюродные братья, матери которых - родные сестры, или дяди и племянники больного по материнской линии и т.д. (рис. 17-4).
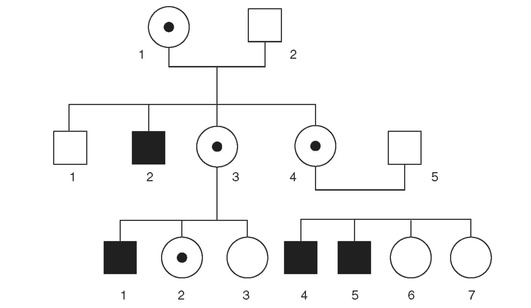
В том случае, если отец болен, то все его сыновья будут здоровы, а все дочери - облигатными гетерозиготными носительницами Х-сцепленного гена. Любые Х-сцепленные рецессивные заболевания значительно чаще поражают мужчин, женщины болеют крайне редко, так как, чтобы заболела женщина, должен болеть ее отец, а мать должна быть гетерозиготой по мутантному гену. Такая ситуация бывает тогда, когда Х-сцепленное рецессивное заболевание не очень резко снижает приспособленность больного. Примером подобного заболевания является Х-сцепленный рецессивный ихтиоз, или цветовая слепота к красному цвету, наследующаяся Х-сцепленно.
Для Х-сцепленных рецессивных, как и для аутосомно-доминантных заболеваний, свойственно возникновение части случаев в результате вновь возникших мутаций. Однако в отличие от аутосомно-доминантных заболеваний, когда при летальности этого заболевания на ранних этапах онтогенеза все его случаи объясняются вновь возникшими мутациями, при Х-сцепленных рецессивных заболеваниях такого не бывает, так как у женщин - гетерозиготных носительниц летального Х-сцепленного рецессивного гена - этот ген не проявляется, поскольку имеется его нормальная копия во второй Х-хромосоме. Еще в 1935 г. Холдейн показал, что при летальном Х-сцепленном заболевании у 1/3 больных его причиной является вновь возникшая мутация, при условии одинаковой частоты мутирования в женских и мужских ПК. В настоящее время показано, что большая часть точковых мутаций в Х-хромосоме возникает во время сперматогенеза. В этом случае доля вновь возникших мутаций, обусловливающих Х-сцепленное рецессивное летальное заболевание (например, миопатию Дюшенна), будет меньше. Данная проблема чрезвычайно важна для медико-генетического консультирования, когда необходимо решить, является ли мать больного ребенка с Х-сцепленным рецессивным летальным заболеванием гетерозиготной носительницей мутантного гена, или это случай вновь возникшей мутации. В первом варианте риск заболевания у ее следующего сына будет равен 50%, а во втором - 0%.
Х-сцепленных доминантных признаков (в том числе и заболеваний) немного. Примером такого заболевания является D-резистентный гипофосфатемический рахит, при котором нарушена резорбция фосфатов в почечных канальцах и, как следствие, - оссификация длинных трубчатых костей, в результате происходит их искривление, что особенно характерно для нижних конечностей. Еще одно Х-сцепленное доминантное заболевание - недержание пигмента (incontinentia pigmenti), проявляющееся нарушением пигментации кожи, а также глазными и неврологическими нарушениями и аномалиями зубов. Особенностью этого заболевания является то, что все гемизиготные мужчины, носители мутантного гена, поражаются настолько сильно, что погибают внутриутробно. В результате только женщины, имеющие это заболевание, попадают в поле зрения врачей, и возникает необходимость отличать Х-сцепленное доминантное наследование от аутосомно-доминантного наследования заболевания, ограниченного полом, такого, например, как РМЖ, обусловленный мутацией в гене BRCA1.
Если мутантный ген отвечает за Х-сцепленное доминантное заболевание, то будут больны 1/2 дочерей и 1/2 сыновей больной матери, т.е., как и при аутосомно-доминантном заболевании, риск заболеть будет одинаковым для детей обоего пола - 1/2, или 50%. В этом случае родословная не позволяет различить два типа наследования. Заподозрить по родословной Х-сцепленное доминантное заболевание позволяет родословная, в которой болен отец. Поскольку отец передает свою Х-хромосому всем дочерям, то при достаточно большом числе больных девочек, а также большом числе здоровых мальчиков в семье предположение об Х-сцепленном доминантном заболевании, наследующемся в этой семье, может быть обоснованным.
Y-сцепленное наследование
В Y-хромосоме найдено небольшое число генов, основная масса которых вовлечена в детерминацию мужского пола или имеет отношение к сперматогенезу. Наследование генов и, соответственно, признаков, сцепленных с Y-хромосомой, называется голандрическим. В этом случае ген (признак) от отца передается только всем сыновьям, но не дочерям.
При Y-сцепленном наследовании ген, локализованный в Y-хромосоме, передается только сыновьям, но не дочерям, т.е. наследование признака происходит исключительно по мужской линии.
Псевдоаутосомное наследование
Этот тип наследования называют еще частично сцепленным с полом. Так наследуются гены, которые расположены в псевдоаутосомных областях Х- и Y-хромосом, т.е. на самых концах коротких плеч этих хромосом. Псевдоаутосомные области Х- и Y-хромосом гомологичны и спариваются в мейозе I. Гены, которые находятся в этих псевдоаутосомных областях, могут за счет кроссинговера переноситься из одной хромосомы в другую. В результате признаки, которые контролируют эти гены, в одной части родословной могут выглядеть как Y-сцепленные, а в другой - как Х-сцепленные. Так наследуется дисхондростеоз, или синдром Лери-Вейля, обусловленный мутациями в гене SHOX, который локализован в псевдоаутосомной области.
Митохондриальное наследование
Митохондриальная ДНК (мтДНК) содержит несколько десятков генов и представлена кольцевой структурой. Особенностью наследования мтДНК является то, что она наследуется только по материнской линии, хотя больными могут быть как мужчины, так и женщины. Это связано с тем, что в яйцеклетке содержится до 200 000 митохондрий, в то время как сперматозоид несет всего несколько митохондрий, да и они могут деградировать в течение сперматогенеза. Мужчины не могут передать митохондриальное заболевание, а его передача потомству больной женщиной не является строго регулярной из-за существования феномена гетероплазмии, т.е. присутствия в митохондриях как ДНК, несущей патогенную мутацию, так и нормальной ДНК. Родословная, в которой наблюдается наследование митохондриального заболевания, отличается от родословной с Х-сцепленным наследованием (хотя в обоих случаях наследование происходит по материнской линии), так как чаще всего больна мать, хотя симптомы заболевания у нее могут быть выражены незначительно, а заболевание может быть передано не только сыновьям, но и дочерям с равной вероятностью.
ОБЩАЯ ФЕНОМЕНОЛОГИЯ ПРОЯВЛЕНИЯ ГЕНОВ МОНОГЕННЫХ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
Предполагается, что в геноме человека содержится примерно 20 000 белок-кодирующих генов. В настоящее время известна последовательность более 16 000 генов, соответственно, последовательность еще нескольких тысяч генов пока неизвестна, но все понимают, что это дело времени, причем не очень продолжительного. На место традиционного метода выявления генов моногенных наследственных болезней с помощью доказательства их типа наследования и последующего генетического картирования, требовавшего большого по объему семейного материала, пришли новые производительные методы сиквенса генома, которые могут быть реализованы на очень небольшом семейном материале, что резко ускорило выявление «новых» генов менделирующих заболеваний.
До настоящего времени считается, что гены, мутации в которых приводят к развитию моногенных заболеваний, в большинстве случаев кодируют полипептидные цепи, или белки. ДНК таких генов составляет приблизительно 1,5% ДНК всего генома. Эту часть генома называют экзомом, а сиквенс экзома является как раз тем подходом, который используется для выявления новых редких генов моногенных заболеваний, о чем сказано ранее.
Диагностика моногенных наследственных болезней, как, впрочем, и других типов наследственных болезней, начинается с их фенотипического описания, затем могут быть использованы биохимические методы диагностики для наследственных болезней обмена веществ или молекулярно-генетические методы, которые являются единственными методами этиологической диагностики этой группы наследственных болезней. Следует, однако, почеркнуть, что фенотипическое описание продолжает оставаться не только важным приемом диагностики, но и основой для выяснения патогенеза моногенных наследственных болезней. Углубленное фенотипическое описание моногенных наследственных болезней получило свое развитие в международной программе Human Phenotype Ontology (http://human-phenotype-ontology.github.io/about.html), раздел которой - Phenomizer может быть использован как подспорье в диагностике и дифференциальной диагностике моногенных наследственных болезней. В этом отношении интересна также серия статей, опубликованных преимущественно в 2009 г. в 149 томе Am. J. Med. Genet. Part A, в которых под общим заголовком «Элементы морфологии» рассматривается рекомендуемая стандартная терминология для аномалий (пороков) развития различных частей тела, таких как нос, губы, уши, руки, ноги и ряд других.
Моногенные наследственные болезни поражают все органы и системы органов человека, нередко поражение затрагивает одновременно различные органы и проявляется в виде синдрома.
Изменения (мутации) далеко не всех генов приводят к развитию моногенных наследственных болезней. Полную и постоянно обновляющуюся информацию о генах, мутации в которых обусловливают развитие наследственных моногенных наследственных болезней, можно найти в таких базах данных, как Online Database of Mendelian Inheritance in Man (OMIM) (https://www.omim.org) или The Human Gene Mutation Database (http://www.biobase-international.com/product/ hgmd). Анализ этих баз данных показывает, что из 16 000 расшифрованных генов человека мутации только примерно 6000 обусловливают возникновение различных моногенных заболеваний и признаков (в соответствующей статистике OMIM на конец апреля 2020 г. было указано, что чуть больше 3800 генов отвечают за моногенные заболевания и нормальные признаки). Это составляет около 20% всех известных генов человека. Наши подсчеты показали, что в OMIM нормальные признаки и заболевания представлены примерно поровну. Предполагается что процент генов, мутации в которых являются причиной моногенных заболеваний, вряд ли сильно изменится, когда будут найдены все гены человека. Одной из возможных причин этого феномена может быть дублирование функций многих генов, особенно так называемых генов «домашнего хозяйства», обеспечивающих поддержание структуры и функции всех клеток организма. Считается, что гены «домашнего хозяйства» составляют большую часть генов генома и активны во всех клетках.
Еще одной причиной того, что мутации не во всех генах проявляются моногенными заболеваниями, может быть ранняя летальность носителей таких мутаций, не выявляемая обычными методами.
О месте проявления мутантных генов, вызывающих моногенные заболевания
Гены по-разному проявляются в различных тканях и органах в соответствии с характером их дифференцировки, которую гены в значительной мере и определяют. Хотя в каждой клетке организма содержится весь набор генов, в клетках с разными типами дифференцировки активны разные гены. Гены гемоглобинов, например, активно работают только в клетках красного ростка крови, и гемоглобины практически невозможно обнаружить в других тканях. Гены миелинов активны преимущественно в клетках периферических нервов, а гены кристаллинов активны в хрусталике. Таких примеров дифференциальной активности генов можно приводить очень много. В сущности, все наследственные болезни, проявляющиеся изолированным поражением отдельных органов, являются такими примерами. Однако есть гены, работающие не в одном органе или ткани, а во многих. В этих случаях гены кодируют белки, или особые РНК, выполняющие какие-либо общебиологические функции. Мутации в таких генах приводят к возникновению наследственных синдромов, причем спектр поражаемых органов и тканей определяется тем, где функция соответствующего гена является очень важной для нормального функционирования этих органов и тканей. Возьмем для примера ауто-сомно-рецессивный синдром атаксии-телеангиэктазии. Клинические проявления этого синдрома разнообразны и включают низкий рост, телеангиэктазии на конъюнктиве глаз и на коже, бронхоэктазы, мозжечковую атаксию, гипогонадизм, гипоплазию тимуса и еще ряд симптомов, в частности, склонность к малигнизации. Синдром обусловлен мутациями в гене АТМ, который картирован на хромосоме 11 в локусе 11q22.3 (OMIM 607585). Ген кодирует белок, который относится к семейству фосфатидилинозитол-3 киназ. Этот белок необходим для репарации повреждений ДНК, так как фосфорилирует ключевые субстраты, участвующие в репарации ДНК и в контроле за протеканием клеточного цикла. С точки зрения функций белка гена АТМ ясны некоторые симптомы синдрома атаксии-телеанги-эктазии, в частности, поражение иммунной системы и склонность к малигниза-ции, так как в этих случаях репарация ДНК от повреждений является критически важным процессом. Вместе с тем другие клинические проявления синдрома, такие как мозжечковая атаксия, гипогонадизм или телеангиэктазии, в настоящее время трудно объяснить, но совершенно очевидно, что контроль за клеточным циклом, в котором участвует белок АТМ, очень важен для нормального формирования и функционирования соответствующих систем.
О времени проявления мутантных генов, вызывающих моногенные заболевания
Еще одной особенностью действия генов, кроме места их действия, является время их действия в течение онтогенеза. Хотя большинство моногенных болезней проявляется достаточно рано в развитии (примерно 75-80%), известно значительное число таких болезней, которые манифестируют в юношеском, взрослом или даже в старческом возрасте. Примером последних является болезнь Альцгеймера, или старческое слабоумие, поздняя форма которой проявляется обычно после 65 лет. Некоторые гены бывают активны в нескольких фазах онтогенеза, в этом случае фенотипические эффекты таких генов могут быть совершенно различны в разных фазах. Особый феномен времени действия генов представляют собой гены гемоглобина. В эмбриональном периоде гемоглобин представлен двумя α- и двумя ε-цепями, у плода гемоглобин представлен двумя α- и двумя γ-цепями, а у взрослых -двумя α- и двумя β-цепями. Кроме того, в А2-гемоглобине две α-цепи соединены с двумя δ-цепями.
К сожалению, наши представления о времени и месте действия всех генов генома человека находятся в зачаточном состоянии. Чтобы его преодолеть, необходимо разработать методы анализа транскриптома в любой момент онтогенеза и в клетках всех тканей.
Распределение генов по хромосомам
К настоящему времени картировано, т.е. установлена локализация, большое число генов наследственных болезней, и для многих из этих генов охарактеризована их функция, т.е. установлено, какие продукты они кодируют (число таких генов превышает уже несколько тысяч). В большинстве случаев это белки, играющие различные роли. Это и структурные белки, и рецепторы, и каналы, и транспортные белки, и ферменты, и т.д. Распределение картированных генов по хромосомам человека неравномерное. Есть хромосомы, в которых локализовано большое число генов, например хромосомы 1, 2, 6, 11, или, напротив, относительно немного генов - 21, 18, Y. Положение картированных генов, в том числе генов моногенных заболеваний, во всех хромосомах человека можно найти в разных источниках, в том числе в OMIM.
Классификации моногенных наследственных болезней
Существует множество классификаций моногенных наследственных болезней, но основная базируется на типах менделевского наследования. Согласно этой классификации, все моногенные наследственные болезни делятся на аутосомные доминантные и рецессивные, Х-сцепленные доминантные и рецессивные и Y-сцепленные заболевания. Некоторые авторы сюда же включают моногенные митохондриальные болезни. В каталоге наследственных признаков человека (OMIM) содержится описание более 5000 аутосомных заболеваний, доминантных и рецессивных примерно поровну, несколько сотен Х-сцепленных заболеваний и относительно немного Y-сцепленных. Кроме того, известно более двух десятков моногенных митохондриальных заболеваний (МЗ), обусловленных мутациями в генах мтДНК.
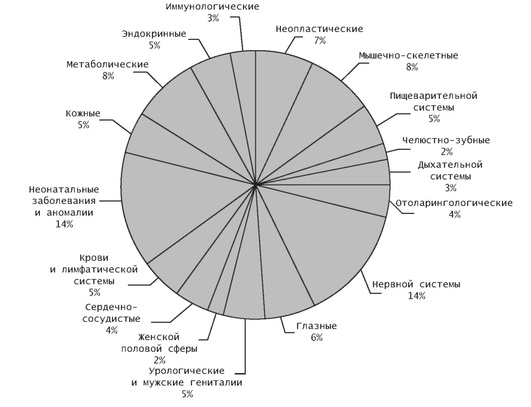
Нередко наследственные болезни классифицируются по тому, поражение какого органа они обусловливают (рис. 17-5). На рис. 17-5 представлен пример такой классификации, построенной на основе анализа проявления 1658 генов, вызывающих преимущественно моногенные заболевания (76% в целом, гены доминантных заболеваний - 30%, рецессивных - 40%, гены, мутации в которых дают как доминантные, так и рецессивные заболевания, - 4%, гены Х-сцепленных заболеваний - 6%). Надо заметить, что это большая часть моногенных болезней, для которых на момент написания статьи был известен молекулярный дефект. Некоторые гены поражают более чем один орган или ткань, но на рисунке эти болезни включены столько раз, сколько органов или тканей поражается. Все поражаемые системы организма представлены 17 категориями, из них две категории - нервная система и неонатальные заболевания - оказываются наиболее нагруженными наследственными болезнями. За ними по частоте следуют наследственные болезни обмена веществ и наследственные болезни, поражающие мышечную систему и скелет. Остальные органы и ткани страдают при наследственных заболеваниях примерно с одинаковой частотой (2-6%). То, что неонатально проявляющиеся наследственные болезни составляют значительную часть всей наследственной патологии, легко объясняется тем, что в раннем развитии любого организма необходима работа большей части генов генома человека. Понятно, что в этом случае значительно возрастают шансы, что мутации большого числа генов будут приводить к различным нарушениям онтогенеза. Известно несколько тысяч наследственных синдромов, проявляющихся, как правило, множественными ПР. Значительная часть этих синдромов наследуется моногенно. Высокая частота наследственных неврологических заболеваний также указывает на то, что в формировании и функционировании нервной системы принимают участие многие гены, и их доля, вероятно, больше, чем доли генов, необходимых для дифференцировки и функционирования других органов.
Основой другой классификации моногенных наследственных болезней является то, какая из молекулярных функций нарушается в случае мутации в соответствующем гене. Число молекулярных функций в исследованиях разных авторов варьирует, но в большинстве случаев к ним относят связывание с нуклеиновыми кислотами, ферментативные, транспортные, моторные, регуляцию транскрипции, сигнальную трансдукцию, структурную, связывание с лигандом, шаперонную, регуляцию активности ферментов и т.д. На рис. 17-6 (см. цв. вклейку) приведены результаты уже цитированного исследования, посвященного классификации генов человека. Для этого предварительно из разных источников была собрана информация о функции 12 221 генов человека, из которых 1430 генов, мутируя, обусловливают развитие заболевания. Следует отметить, что авторы цитируемого исследования используют χ2 и другие статистические показатели для подтверждения надежности своих заключений. При сравнении этих двух групп генов выявляется, что моногенные заболевания возникают при мутациях генов всех функциональных классов. Однако представленность генов, вызывающих моногенные заболевания, в ряде случаев не совпадает с представленностью генов соответствующего функционального класса в общем пуле известных генов. Так, моногенные заболевания чаще встречаются при мутациях в генах ферментов, белков-транспортеров и структурных белков. В то же время гены белков, связывающихся с нуклеиновыми кислотами, среди тех, что вызывают наследственные болезни, оказываются недопредставленными.
Еще одна классификация наследственных болезней основана на анализе того, какой биологический процесс нарушается при мутации в соответствующем гене. Как и в предыдущем случае, разные авторы по-разному классифицируют биологические процессы. Один из типов классификации включает следующие биологические процессы: транспорт, ответ на стресс, клеточный цикл, процессы развития, метаболизм, подвижность клеток, межклеточные сообщения, физиологические процессы, смерть. При такой классификации биологических процессов оказывается, что гены наследственных заболеваний чаще, чем ожидается, представлены в ответе на стресс и клеточной подвижности, а гены, продукты которых осуществляют коммуникацию между клетками, среди тех, что вызывают наследственные моногенные болезни, недопредставлены (рис. 17-7, см. цв. вклейку).
Следует оговориться, что результаты распределения наследственных болезней в рамках рассмотренных классификаций можно считать предварительными, или условными. Они могут измениться по мере включения в анализ вновь изученных генов.
Чаще, чем ожидается, оказывается, что гены, продукты которых участвуют в выполнений таких молекулярных функций, как ферментативная и транспортная, наследуются аутосомно-рецессивно, а гены, участвующие в регуляции транскрипции, кодирующие структурные белки или белки сигнальной трансдукции, наследуются аутосомно-доминантно. Мутации в генах, участвующих в таких биологических процессах, как метаболизм и стресс, чаще приводят к рецессивным заболеваниям, а участвующих в контроле клеточного цикла, процессах развития или клеточной коммуникации - обусловливают доминантные заболевания.
Типы мутаций в генах, вызывающих моногенные заболевания
Моногенные наследственные болезни обычно вызываются мутацией в гене, затрагивающей относительно небольшие отрезки ДНК. Наиболее частыми являются точковые мутации. Точковые мутации затрагивают один нуклеотид, и если при этом пуриновое основание остается пуриновым, а пиримидиновое - пиримидиновым, то такие замещения называются транзициями, в противном случае замещения называются трансверсиями. В зависимости от того, как точковые мутации изменяют белковый продукт гена, их классифицируют на молчащие, миссенс-, нонсенс-, регуляторные и мутации, нарушающие процессинг РНК. Молчащие мутации обязаны вырожденности генетического кода. Например, валин кодируется следующими кодонами: GUU, GUA, GUC и GUG, и поэтому любое замещение третьего нуклеотида будет молчащей заменой. Миссенс-мутации обусловливают замещение одной аминокислоты на другую. Эти замещения могут приводить к изменению функции белка, и в этом случае можно ожидать развития патологии, а могут и не приводить к таким последствиям. Миссенс-мутации являются самыми частыми из точковых мутаций. Нонсенс-мутации приводят к возникновению стоп-кодона (UAA, UAG и UGA) в необычном месте гена, что обусловливает преждевременное прекращение трансляции. Обычно такая мРНК оказывается нестабильной и подвергается разрушению до начала трансляции. Регуляторные мутации обычно возникают в промоторе и других регулирующих активность гена элементах, таких как энхансеры, сайленсеры, или локус-контролирующих областях. Обычно мутации такого сорта приводят к снижению продукции мРНК, но в некоторых случаях, напротив, увеличивают уровень транскрипции (примером является регуляторная мутация при наследственной персистенции фетального гемоглобина). Мутации, нарушающие процессинг мРНК, либо изменяют сайт сплайсинга мРНК, либо нарушают 5'-кэппирование или З'-полиаденилирование. Мутации сайта сплайсинга являются достаточно частой причиной наследственной патологии (например, мутация сайта сплайсинга в гене TCIRG1 при рецессивном летальном остеопе-трозе, изученном нами в Чувашии). Кроме точковых мутаций, к развитию моногенных заболеваний могут приводить и более обширные изменения нуклеотидной последовательности гена, такие как делеции, дупликации, инсерции и другие.
Мутации, приводящие к развитию моногенного заболевания, могут быть классифицированы как мутации, ведущие к потере функции, и мутации, ведущие к приобретению функции. Первый тип мутаций характерен для рецессивных заболеваний. В норме продукты рецессивных генов синтезируются избыточно. Для многих наследственных болезней обмена веществ, которые наследуются рецессивно, типичной оказывается ситуация, когда сохранение активности мутантного фермента на уровне 10%, а иногда и ниже по сравнению с нормой, оказывается достаточным, чтобы патологический фенотип был выражен слабо или даже не проявлялся. В то же время если доминантное заболевание обусловлено мутацией, приводящей к потере функции, то это означает, что одной дозы гена и, следовательно, половины продукта этого гена явно недостаточно для его нормального функционирования. Этот феномен называется гаплонедостаточностью.
Мутации, ведущие к приобретению функции, проявляются либо повышенной продукцией белка, кодируемого соответствующим геном, либо даже изменением качества этого белка. Такие мутации встречаются значительно реже, чем мутации, приводящие к потере функции. Наиболее яркими примерами мутаций, ведущих к приобретению функций, являются мутации в гене гентингтина, вызывающие хорею Гентингтона. В этом случае образуется избыточное количество белка, который формирует агрегаты в клетках, обладающие нейротоксическим эффектом. При ахондроплазии приобретение функции связано с тем, что возрастает активность рецептора фактора роста фибробластов (мутация в гене FGFR3, OMIM 134934), что ведет в конечном счете к торможению роста костей. Что касается появления белка с новыми качествами, то примером этого являются некоторые мутации в генах соматических клеток, которые приводят к образованию так называемых химерных генов, образующих новый белок, что наблюдается при некоторых формах рака.
Многие белки функционируют как димеры или даже мультимеры, и такие функциональные единицы могут быть продуктом не одного, а двух и более генов. В этом случае мутация одного гена может приводить к нарушению функционирования димера или мультимера, и таким образом проявляется доминантно-негативный эффект, т.е. нарушение функции одного гена мешает проявлению других генов. Многие структурные белки, ТФ, сигнальные белки и белки, связывающиеся с нуклеиновыми кислотами, функционируют в составе мультимерных структур, поэтому они наиболее чувствительны к доминантно-негативному эффекту. Может быть, наиболее показателен в этом отношении коллаген, образующий мультимерную структуру. Мутации в генах разных цепей коллагена обнаруживают четкий доминантно-негативный эффект, выявляющийся при таких наследственных болезнях, как доминантные формы синдрома Элерса-Данло (EDS), при которых наблюдается поражение разных производных соединительной ткани, а также доминантные формы несовершенного остеогенеза, при котором нарушается косте-образование, и многих других наследственных болезнях.
Локусная генетическая гетерогенность моногенных наследственных болезней
На примере коллагенопатий, к числу которых относится EDS, будет рассмотрен такой феномен, как локусная генетическая гетерогенность наследственных болезней. Под локусной генетической гетерогенностью понимают феномен, когда сходные фенотипы являются результатом проявления разных генов, кодирующих сходные по своей функции белки. Локусная генетическая гетерогенность является естественным инструментом изучения генетической регуляции элементарных морфологических и физиологических, в широком смысле этого понятия, признаков. Локусная генетическая гетерогенность обнаруживает отдельные геноконтролируемые звенья в цепи становления таких признаков и позволяет установить их порядок. Именно такой подход был использован Бидлом и Тэтумом при создании биохимической генетики на Neurospora crassa в 40-50-х годах XX столетия.
Коллаген, основной белок соединительной ткани, представляет собой фибриллярный белок, основная форма которого, так называемый тропоколлаген, представляет собой самособирающуюся спираль, образованную тремя α-цепями (в настоящее время описано более 25 вариантов этой цепи). Известно более 19 форм, или типов, коллагена, отличающихся по первичной структуре полипептидных цепей, функциям и локализации. Наиболее распространенными являются первые три типа коллагена, составляющие до 95% всего коллагена организма. Типы коллагена принято обозначать римскими цифрами: I, II, III, IV и т.д. Гены коллагенов записываются арабскими цифрами, например, COL1 - ген коллагена I типа, к этому символу приписываются буква А (обозначает α-цепь) и арабская цифра (обозначает вид α-цепи); например, COL1A1 и COL1A2 - α1 и а2 -цепи коллагена I типа.
Хотя гены коллагенов всех типов имеются в каждой клетке всех тканей и органов, коллагены разных типов по-разному представлены в разных тканях, что отражает в них дифференциальную активность разных генов коллагена. Это означает, что в одних тканях более активны одни гены коллагенов, а в других - другие. Так, коллаген I типа (гены COL1A1 и COL1A2 - OMIM 120150, 120160) наиболее представлен в коже, сухожилиях, кости, роговице, плаценте, артериях, печени и дентине, коллаген II типа (ген COL2A1 - OMIM 120140) - в хряще, межпозвоночных дисках, стекловидном теле и роговице, коллаген III типа (ген COL3A1 - OMIM 120180) - в артериях, матке, коже плода и строме паренхиматозных органов, коллаген IV типа (гены COL4A1-COL4A6 - OMIM 120130, 120190, 120070, 303361, 120131) в базальных мембранах, коллаген V типа (гены COL5A1-COL5A3 - OMIM 120215, 120216, 120190) - в коже, роговице, кости, хрящах, межпозвоночных дисках и плаценте, коллаген VI типа (гены COL6A1-COL6A3 - OMIM 120220, 120240, 120250) в хрящах, кровеносных сосудах, связках, коже, матке, легких и почках и т.д. Такая разная представленность разных форм коллагена в тканях и, соответственно, разная активность генов коллагена во многом объясняют разнообразие клинических форм EDS и причину генетической гетерогенности этого синдрома. По классификации Villefranche выделяют 6 основных типов синдрома:
При EDS I наиболее заметны изменения кожи и гипермобильность как крупных, так и мелких суставов, нередко наблюдаются скелетные деформации и другие ортопедические изменения. В 50% случаев наблюдаются преждевременные роды. Разрывы аорты или кишечника редки. Для EDS II характерны те же признаки, что и для EDS I, но выраженные в меньшей степени.
Генетический анализ EDS I показал, что эта клиническая форма является генетически гетерогенной. Установлено, что EDS I может быть обусловлен мутациями в гене COL5A1, картированном на хромосоме 9q34, или в гене COL5A2, картированном на хромосоме 2q31. Вероятно, мутации в других генах также могут обусловить проявления EDS I. Напомним, что коллаген V типа наиболее представлен в коже, роговице, кости, хрящах, межпозвоночных дисках и плаценте, поэтому мутации в генах коллагена V типа объясняют основные клинические проявления EDS I.
Синдром EDS II обусловлен мутациями в тех же генах, что и при EDS I. Возможно, более легкие клинические проявления при EDS II обусловлены вторым типом генетической гетерогенности, так называемой аллельной гетерогенностью, когда разные мутации в одном и том же гене могут обусловить разные клинические проявления заболевания или даже возникновение клинически иного заболевания. Об этом типе генетической гетерогенности еще будет сказано далее.
EDS IV называют сосудистым типом из-за нередко возникающих разрывов крупных артерий и кишечника. В то же время поражения кожи суставов при этом типе синдрома менее выражены. В частности, гипермобильность может наблюдаться только для межфаланговых суставов. Кожа, хотя и не так растяжима, как при EDS I и II, но очень ранима, и при заживлении образуются типичные «папиросные» рубцы. Причиной EDS IV, как правило, являются мутации в гене COL3A1, который обнаруживает высокую активность в артериях, матке, коже плода и строме паренхиматозных органов. Т.е. в этом случае, как и при EDS I и II, поражается та ткань или орган, где мутантный ген обнаруживает высокую активность.
Выделяют две формы EDS VII типа - А и В. Клинически эти формы плохо различимы, и основная симптоматика касается суставов. У больных с этим типом синдрома часто наблюдаются двусторонний врожденный вывих бедра, подвывихи в других крупных суставах. В то же время изменения кожи выражены незначительно. Причиной этих двух форм синдрома являются мутации в генах COL1A1 и COL1A2 соответственно. Мутации, вызывающие этот синдром, изменяют в α-1-и α-2-цепях коллагена I типа сайт, который расщепляется протеазой. В результате тип I проколлагена не может превратиться в коллаген. Наиболее драматичным эффектом действия этих двух мутантных генов являются изменения в хрящевых поверхностях суставов, но спектр клинических проявлений синдрома значительно шире и может включать паховую грыжу, умеренную гиперрастяжимость кожи, скелетные изменения, в частности, сколиоз и деформацию грудной клетки, низкий рост и другие симптомы.
Продолжим обсуждение проблемы локусной генетической гетерогенности на примере несовершенного остеогенеза, ряд форм которого, как и при EDS, связан с мутациями в генах коллагена.
Несовершенный остеогенез I типа наследуется аутосомно-доминантно. Рост больных страдает незначительно, у взрослых возникает тугоухость - либо нейросенсорная, либо проводящая, наблюдается пролапс митрального клапана, переломы длинных трубчатых костей впервые возникают, когда ребенок начинает ходить, число их варьирует, после наступления половой зрелости частота переломов уменьшается, но затем вновь возрастает у пожилых, кожа тонкая, легко ранимая, склеры голубые. Мутации обнаруживаются обычно в гене α-1-цепи коллагена I типа (COL1A1).
Несовершенный остеогенез IIa типа также наследуется аутосомно-доминантно. Клинические проявления этого типа несовершенного остеогенеза значительно более тяжелые, чем I типа: дети рождаются с низким весом, наблюдается водянка плода, рост резко снижен, наблюдаются многочисленные врожденные переломы, сердечная и легочная недостаточность, склеры голубые, кожа тонкая, ранимая, нос клювовидный. Обычно дети с этим синдромом погибают до года. В то же время несовершенный остеогенез IIa типа может быть обусловлен мутациями в гене α-1-цепи коллагена I типа (COL1A1), как и несовершенный остеогенез I типа. Правда, при несовершенном остеогенезе IIa типа нередко мутации находят в гене α-2-цепи коллагена I типа.
Несовершенный остеогенез III типа может наследоваться как аутосомно-доминантно, так и аутосомно-рецессивно. Карликовость, как и множественные переломы длинных трубчатых костей, обнаруживаются при рождении, лицо у больных имеет треугольную форму, лоб нависает, заметна микрогнатия. У больных наблюдается несовершенный дентиногенез как молочных, так и постоянных зубов, развивается тугоухость. Характерно базилярное вдавление. При этой клинической форме мутации наблюдаются в тех же генах, что и при несовершенном остеогенезе IIa типа, т.е. в генах COL1A1 и COL1A2.
Несовершенный остеогенез IV типа наследуется аутосомно-доминантно. По клиническим проявлениям это более легкая форма, чем предыдущая: рост больных ниже 5 перцентилей, нередко развивается отосклероз и наблюдается тугоухость, число переломов варьирует, хотя они могут возникать и внутриутробно, после наступления половой зрелости их частота снижается, голубые склеры наблюдаются редко. Несовершенный дентиногенез также характерен не для всех больных. Мутации обнаруживаются в генах COL1A1 и COL1A2.
Итак, с генетической точки зрения все клинические формы несовершенного остеогенеза можно расценивать как одну нозологическую форму, поскольку их причиной являются одни и те же мутантные гены. Ситуация с этими генами коллагена (COL1A1 и COL1A2) в действительности выглядит еще более сложной, если обратить внимание на то, что две формы EDS VII типа - А и В - также обусловлены мутациями в генах COL1A1 и COL1A2. Природа генетической гетерогенности несовершенного остеогенеза отлична от той, что продемонстрирована для EDS. При несовершенном остеогенезе разные мутации в одних и тех же генах дают разные клинические фенотипы. Такой тип генетической гетерогенности называется аллельной генетической гетерогенностью, и мы еще вернемся к нему далее.
Рассмотрим еще несколько состояний, которые можно отнести к наследственным коллагенопатиям. Это позволит нам еще раз подтвердить положение о том, что патология проявляется в тех органах и тканях, где соответствующие гены проявляют высокую активность. Различают несколько форм синдрома Альпорта, прежде всего по типу наследования. Наиболее частой является Х-сцепленная рецессивная форма, следующая за ней по частоте - аутосомно-доминантная и наиболее редкая - рецессивная форма синдрома. Для Х-сцепленной формы, кроме проявлений симптомов наследственного нефрита (микроскопическая гематурия, протеинурия, цилиндрурия, лейкоцитурия), характерны также нейросенсорная тугоухость, иногда поражение глаз (лентиконус, сферофакия, врожденная катаракта), тромбоцитопения и аномалия Мая-Хеглина, ихтиоз, гипопаратиреоз, диффузный лейомиоматоз. Заболевание обусловлено мутациями в гене α-5-цепи коллагена 4-го типа (COL4A5). Гены этого типа коллагена проявляются в базальной мембране почечных канальцев, и с этой точки зрения другие плейотропные (множественные) эффекты мутантного гена не вполне понятны, в частности, поражение органа слуха и глаз, а также ихтиоз.
Плейотропные эффекты генов моногенных заболеваний
Сейчас мы использовали впервые термин «плейотропные эффекты гена». Следует напомнить, что различают два типа плейотропных эффектов гена. Один из них называется первичной плейотропией и обусловлен тем, что ген независимо действует в разных тканях, но, так как в клетках этих тканей наборы активных генов могут быть разными, то и проявления исследуемого гена также могут быть разными, так как его продукты взаимодействуют с продуктами разных генов. Вторичная плейотропия объясняется патологическим процессом, который инициируется мутантным геном и представляет собой цепь взаимосвязанных патофизиологических событий. Плейотропия является, таким образом, одним из фундаментальных особенностей действия генов. Рассмотренные ранее коллагенопатии, такие как EDS и несовершенный остеогенез, демонстрируют практически для каждой отдельной нозологической формы плейотропные проявления действия мутантных генов. В большинстве случаев плейотропное действие генов при этих двух наследственных заболеваниях может быть отнесено к первичной плейотропии, связанной с независимым проявлением генов в разных тканях, но деформации скелета, как и сами переломы длинных трубчатых костей и даже тугоухость, можно рассматривать как проявление вторичной плейотропии. Сходным образом в случае синдрома Альпорта поражение почек является первичным событием, а развивающиеся гематурия, протеинурия, гипертония и некоторые другие симптомы - проявлением вторичной плейотропии.
Для доминантной формы синдрома Альпорта также характерны нефрит, нефротический синдром, проявляющийся гематурией, гипофосфатемией, нефрокальцинозом, протеинурией, азотемией и, как при Х-сцепленной форме, поражением органа слуха (нейросенсорная тугоухость) и глаз (лентиконус, передняя полярная катаракта, миопия). Доминантная форма синдрома обусловлена мутациями в гене α-3-цепей коллагена IV типа. То, что в обеих формах синдрома, кроме почек, поражаются глаза и уши, однозначно свидетельствует об участии коллагена IV типа в поддержании структур этих органов. Рецессивная форма синдрома Альпорта также проявляется нефритом, симптомом которого является гематурия. Кроме того, у больных с этой формой наблюдается тугоухость. Рецессивный синдром Альпорта обусловлен гомозиготностью или компаунд-гетерозиготностью (напоминаем, что это означает, что родители больного несут разные мутации в одном гене) по мутациям в генах α-3 или α-4 коллагена IV типа. Оба гена расположены в хромосоме 2q.
Еще одна форма наследственных коллагенопатий - это синдром Стиклера. Стиклер описал этот синдром, практикуя в клинике Мейо, на основе анализа большой родословной как аутосомно-доминантное состояние, включающее прогрессирующую миопию, которая начинается на первом десятилетии жизни, и часто ведет к отслойке сетчатки и слепоте. У больных обнаруживаются также преждевременные дегенеративные изменения в суставах и ряд других клинических признаков, в частности, марфаноподобный внешний вид, нейросенсорная тугоухость, лицевые аномалии, расщелина неба, скелетные аномалии в целом можно характеризовать как умеренную спондилоэпифизарную дисплазию. Синдром Стиклера I типа обусловлен мутациями в α-1-цепи коллагена II типа (COLIIA1). Во многих отношениях сходная клиническая картина найдена при синдроме Стиклера II типа, при котором мутации обнаружены в гене α-1-цепи коллагена XI типа (COLXIA1).
Мутации в этом же гене являются также причиной синдрома Маршалла, который по ряду признаков сходен с синдромом Стиклера, в частности, по гипоплазии средней части лица, миопии, нейросенсорной тугоухости, но по ряду признаков четко выделяется в самостоятельный синдром: низкому росту, утолщению костей черепа, отсутствию фронтальных синусов, короткому носу, толстым губам, ряду скелетных деформаций. Синдром Стиклера III типа отличается от двух предыдущих форм синдрома отсутствием глазной симптоматики. Он обусловлен мутациями в гене α-2-полипептидной цепи коллагена XI типа (COLXIA2).
Проблема плейотропного действия генов чрезвычайно важна для понимания патогенеза такой обширной группы моногенных заболеваний, как синдромы МВПР. В существующих базах данных, таких как POSSUM (Австралия), СИНГЕН (Белоруссия), London Dysmorphology Data Base (Англия), или таких руководствах, как «Наследственные синдромы» по Дэвиду Смиту или «Наследственные синдромы и медико-генетическое консультирование» С.И. Козловой и Н.С. Демиковой, содержатся сведения о многих сотнях таких синдромов. Их диагностика и понимание патогенеза не могут быть полноценными без знания, какую роль играют первичная и вторичная плейотропия в клинических проявлениях синдромов МВПР.
Мы уже коснулись проблемы аллельной генетической гетерогенности, при которой мутации в одном гене могут приводить к проявлению настолько разных фенотипов, что они клинически выделяются в отдельные нозологические формы с характерной для них клиникой (синдром Элерса-Данло VII типа и несовершенный остеогенез, различные формы несовершенного остеогенеза).
Может быть, одним из самых ярких примеров аллельной генетической гетерогенности являются наследственные ламинопатии. Ламины представляют собой структурные белки, которые являются компонентами ядерной ламины, представляющей собой белковую сеть, которая выстилает внутреннюю оболочку ядра клетки. Эта оболочка определяет форму ядра и его размеры. Ламины относятся к классу промежуточных филаментов (рис. 17-8).
В клетках млекопитающих описаны три типа ламинов - А, В и С. Ген лами-нов А/С (LMNA) картирован на хромосоме 1 (1q21.2). Его кодирующая часть занимает 24 т.п.н. и состоит из 12 экзонов. АС в 10 экзоне обусловливает образование 2 типов мРНК, которые кодируют преламин А и ламин С. Ламины играют важную роль в поддержании взаимодействия между ядром и цитоскелетом клетки и, по-видимому, во многих других клеточных процессах, включая дифференци-ровку. Мутации в гене ламина А/С обусловливают возникновение разнообразной наследственной патологии (установлено, что они являются причиной по крайней мере 10 наследственных синдромов). Наследственные болезни, обусловленные мутациями в гене ламина А/С, могут быть классифицированы на 4 основных типа: болезни поперечно-полосатой мускулатуры и сердечной мышцы, синдромы липодистрофий, периферические нейропатии и синдромы преждевременного старения. К первой группе относятся мышечная дистрофия Эймери-Дрейфуса, поясно-конечностная мышечная дистрофия типа 1В, врожденная мышечная дистрофия, дилатационная кардиомиопатия и синдромы наследственных дефектов сердечной проводимости. Синдром липодистрофии включают частичную липодистрофию 2-го типа, при которой наблюдаются региональная липодистрофия с прогрессирующей дегенерацией адипоцитов, резистентность к инсулину и сахарный диабет, признаки мышечной дистрофии. Мутации в гене ламина А/С в некоторых случаях обусловливают невральную амиотрофию (болезнь Шарко-Мари-Тута, тип 2В1). Синдромы преждевременного старения, обусловленные мутациями в гене ламина А/С, прежде всего представлены аутосомно-доминантным синдромом прогерии Хатчинсона-Гилфорда, который проявляется рано развитием гипоплазии средней части лица, постнатальной задержкой роста, отсутствием подкожного жирового слоя, ранней ИБС, инфарктами миокарда и некоторыми другими признаками. Нередко мутации в этом гене находят и при синдроме Вернера, еще одном из синдромов прогерии, и при других прогероидных синдромах. Мутации в гене ламина приводят также к возникновению рестриктивной дерматопатии, рецессивного летального синдрома, проявляющегося внутриутробной задержкой роста, пороками сердца, гипоплазией легких, кифосколиозом, контрактурами суставов, ригидной кожей и ее эрозиями, гипоплазией надпочечников, особенным лицом и рядом других клинических симптомов. Корреляционный анализ показал, что существует заметная связь между фенотипом и местом, в котором произошла мутация гена. Иными словами, если ген работает во многих тканях, то разные домены белка, который он кодирует, имеют в этих тканях разную функциональную значимость. Кроме того, как мы уже указывали, даже если ген кодирует только один продукт, то в разных тканях он взаимодействует с продуктами разных генов, и вследствие этого конечные фенотипические признаки, зависящие от этого гена, в разных тканях могут быть совершенно различными. Это заключение имеет отношение к объяснению природы аллельной генетической гетерогенности в целом.
ЭКЗОНЫ ГЕНА ЛАМИНА А/С
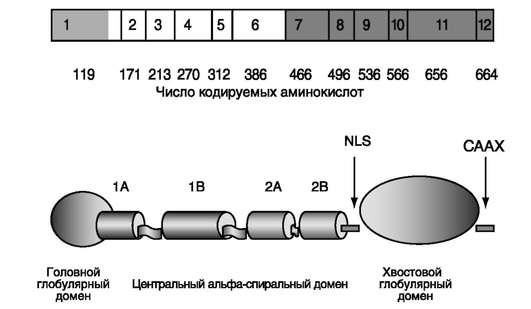
Рассмотренные нами наследственные болезни представляют собой замечательный пример особенностей проявления генов, кодирующих структурные белки, которые заключаются в том, что гены характеризуются определенным местом своего действия. Что касается времени действия гена, то это важная характеристика действия мутантного гена, которая учитывается в медико-генетическом консультировании при расчете риска повторного возникновения или проявления возрастзависимого заболевания.
Пенетрантность и экспрессивность генов моногенных заболеваний
Проявление мутантных генов описывается также такими феноменами, как пенетрантность и экспрессивность. Оба термина были предложены Н.В. Тимофеевым-Ресовским в 1925 г. Первый из этих феноменов означает долю особей, носителей соответствующего мутантного гена, у которых эти гены проявляются. Эта доля может варьировать от 100%, когда ген проявляется у всех носителей, до значений, близких к 0, когда, несмотря на носительство, проявлений гена не обнаруживается. В случае низкой пенетрантности того или иного гена возникает естественный вопрос о том, насколько велика роль этого гена в проявлении наследственного заболевания - фундаментальный вопрос в генетике сложно наследуемых заболеваний. Неполная пенетрантность особенно типична для доминантных заболеваний. В родословной неполная пенетрантность проявляется так называемым пропуском поколения.
Примером генов с неполной пенетрантностью является ген BRCA1, мутации в котором обусловливают возникновение РМЖ и яичников. Установлено, что пенетрантность этого гена составляет около 80%, т.е. риск заболеть для носителей мутантного гена в течение жизни составляет около 80%. Пенетрантность, как и возраст проявления заболевания, учитывается при расчете повторного риска в медико-генетическом консультировании.
Экспрессивность мутантных генов означает разную степень проявления генов, что с медицинской точки зрения означает более тяжелое или более легкое течение наследственного заболевания. Варьирующая экспрессивность характерна для многих, если не всех, наследственных болезней, но наиболее отчетливо она проявляется для доминантных заболеваний.
Антиципация - один из феноменов проявления моногенных заболеваний
Еще один феномен, требующий освещения в этой главе, - это феномен антиципации. Под антиципацией понимают более раннее и/или тяжелое проявление симптомов наследственного заболевания у детей по сравнению с больным родителем. Хотя существование антиципации предполагалось в генетике достаточно давно, но многие известные генетики считали, что это статистический артефакт, обусловленный особенностями сбора семей для анализа. Только относительно недавно был найден и проанализирован генетический механизм антиципации. Оказалось, что антиципация наблюдается достаточно часто при наследственных заболеваниях, обусловленных особым, новым типом мутаций, связанных с так называемым расширением зоны тринуклеотидных повторов. Этот тип мутаций называют еще динамическими мутациями. Тринуклеотидные повторы встречаются в генах нередко. Они найдены в экзонах, интронах, в 5'- и З'-нетранслируемых областях генов. Как следует из названия, тринуклеотидные повторы представляют собой повторяющиеся последовательности из трех нуклеотидов (например, CGG). Во многих случаях эти повторы не имеют каких-либо специфических проявлений, т.е. бессимптомны. Однако в некоторых генах эти повторы во время мейоза могут расширяться, т.е. число повторов увеличивается, что приводит к появлению наследственного заболевания. При этом число тринуклеотидных повторов должно превысить некий численный порог, разный для разных генов и, соответственно, заболеваний. Так, в гене миотонической дистрофии в норме содержится от 5 до 30 CTG повторов в З'-нетранслируемой области гена DMPK (OMIM 605377). Если число повторов увеличивается до 50, могут появиться отдельные симптомы заболевания. При числе повторов, превышающем 100, миотоническая дистрофия начинает проявлять раньше, но все же у взрослых. При числе повторов 400 и больше заболевание начинает проявляться в детстве. Механизм, обусловливающий увеличение числа повторов в мейозе, остается неясным. В настоящее время феномен антиципации - хорошо установленный факт для целого ряда неврологических заболеваний, таких как хорея Гентингтона, миотоническая дистрофия, некоторые формы спиноцеребеллярных атаксий (СЦА) (все наследуются аутосомно-доминантно), синдром ломкой Х-хромосомы (наследуется Х-сцепленно), атаксия Фридрейха (наследуется аутосомно-рецессивно). Если тринуклеотидные повторы располагаются в белок-кодирующей части гена, то в результате их расширения образуется мутантный белок с приобретенной функцией. Это характерно, например, для хореи Гентингтона, когда расширение зоны тринуклеотидных повторов CAG приводит к удлинению полиглутаминового тракта в белке, который называют гентингтином, о чем уже упоминалось в этой главе. Если повтор присутствует в нетранслируемой области гена, то его расширение может повлиять на экспрессию гена, как это происходит при синдроме ломкой Х-хромосомы. Кроме тринуклеотидных повторов, расширение которых ведет к наследственной патологии, известны также наследственные болезни, обусловленные расширением зоны тетрануклеотидных повторов (CCTG при миотонической дистрофии 2-го типа) и пентануклеотидных повторов (АТТСТ при СЦА 10-го типа). Более подробно описание и анализ наследственных болезней, обусловленных расширением зоны тринуклеотидных повторов, представлены в одной из глав этого руководства.
Особенности проявления генов, мутации в которых обусловливают наследственные болезни обмена веществ
Проявление генов структурных белков обычно наблюдается в тех тканях и органах, где они активны. Мы отмечали это раньше. Этот феномен носит название автономного действия гена - термин, который редко используется в медицинской генетике. Однако это нехарактерно для генов наследственных болезней обмена веществ. В случае некоторых наследственных болезней обмена веществ можно говорить о неавтономном проявлении гена, когда продукты гена, в частности различные метаболиты, попадают в биологические жидкости и вызывают патологические эффекты в различных тканях и органах.
Обмен веществ у человека представляет собой строго скоординированное взаимодействие большого числа метаболических путей. Мы различаем обмен углеводов, липидов, аминокислот, мукополисахаридов и т.д., которые состоят из цепей последовательных реакций превращения определенного субстрата, где каждая реакция осуществляется специальным ферментом. В метаболизме играют важную роль внутриклеточные органеллы, такие, например, как митохондрии, лизосомы, или пероксисомы, белки, обеспечивающие транспорт внутри клеток, рецепторы, расположенные на поверхности клеток, и другие белки. Наследственные болезни обмена веществ насчитывают сотни нозологических единиц, что в определенной степени отражает сложность генетического контроля метаболизма. Как уже отмечалось, большинство наследственных болезней обмена наследуется аутосомно-рецессивно либо Х-сцепленно рецессивно, а мутации в генах, которые их обусловливают, относятся к мутациям, приводящим к утрате функции. При наследственных дефектах ферментов, многие из которых синтезируются в клетках печени, иногда невозможно предсказать, какая ткань или орган будут поражены. В качестве примера можно привести ФКУ, при которой в большей степени, чем остальные органы, страдают мозг и нервная система, и до сих пор обсуждается вопрос, является ли причиной этого накопление в крови фенилаланина и промежуточных продуктов его метаболизма. Сходным образом трудно объяснить симптомы такого наследственного заболевания, как болезнь, при которой моча имеет запах кленового сиропа. Это рецессивное заболевание связано с дефектом мультимерного ферментативного комплекса, известного как декарбоксилаза α-кетокислот с разветвленной цепью. Комплекс кодируется по крайней мере четырьмя генами и участвует в метаболизме таких аминокислот, как валин, лейцин и изолейцин. В случае мутации в любом из генов в крови и моче повышается уровень этих аминокислот, и моча начинает пахнуть кленовым сиропом. У младенцев с этим заболеванием наблюдаются дегидратация, анорексия и апатия, затем развиваются судороги и спастика. В отсутствие лечения нарастает коматозное состояние и наступает смерть. При наследственных болезнях обмена веществ, когда возникает ферментативный блок на пути превращения того или иного субстрата, патогенетически значимыми оказываются накапливающиеся метаболиты и продукты их альтернативных превращений. Именно поэтому трудно предсказать, для каких тканей и органов эти накапливающиеся метаболиты будут токсическими, но чаще всего ими поражаются головной мозг и нервная система. Заметим, что наследственным болезням обмена веществ в этом руководстве посвящены две главы.
В настоящей главе мы постарались осветить общие проблемы, касающиеся моногенных наследственных болезней, феноменологию проявления мутантных генов, вызывающих эти заболевания. Хотя мы приводили примеры ряда наследственных болезней, коротко описывая их клинические проявления и возможный молекулярный патогенез, но главным было дать представление читателям о том, что характеризует действие мутантных генов, обусловливающих формирование вместе с другими генами, а также факторами окружающей и внутренней среды сложной картины клинического проявления моногенных наследственных болезней.