Эндокринология : национальное руководство / под ред. И. И. Дедова, Г. А. Мельниченко. - 2-е изд. , перераб. и доп. - Москва : ГЭОТАР-Медиа, 2021. - 1112 с. : ил. - 1112 с. - ISBN 978-5-9704-6054-2. |
Аннотация
Национальные руководства - серия практических руководств по основным медицинским специальностям, включающих специальную информацию, необходимую врачу для непрерывного последипломного образования. В отличие от других изданий в национальных руководствах равное внимание уделено профилактике, диагностике, фармакотерапии и немедикаментозным методам лечения.
В национальном руководстве "Эндокринология" приведены современные рекомендации по профилактике, диагностике, лечению эндокринных заболеваний и реабилитации эндокринологических больных. Особое внимание уделено ведению больных с наиболее распространенными заболеваниями эндокринной системы, такими как сахарный диабет, ожирение, остеопороз, болезни щитовидной железы. Рекомендации по диагностике, лечению и профилактике эндокринных заболеваний подготовлены ведущими специалистами и отражают объединенную, согласованную позицию отечественной научной школы.
В настоящем, втором издании пересмотрены и обновлены все разделы руководства с учетом последних международных и отечественных рекомендаций.
Руководство предназначено эндокринологам, терапевтам, врачам общей практики, а также студентам старших курсов медицинских вузов, интернам, ординаторам, аспирантам.
Глава 14. Нейроэндокринные заболевания
ИНЦИДЕНТАЛОМА ГИПОФИЗА
Иловайская И.А.
СИНОНИМЫ
«Болезнь современных технологий».
ОПРЕДЕЛЕНИЕ
Инциденталома гипофиза (от англ. incidental - случайный) - объемное образование гипофиза, случайно выявленное при МРТ или КТ, не сопровождаемое явными клиническими симптомами нарушения гормональной секреции.
НЕОТЛОЖНЫЕ МЕРОПРИЯТИЯ ПО ДИАГНОСТИКЕ И ЛЕЧЕНИЮ
Не применяют.
ЭПИДЕМИОЛОГИЯ
Частота встречаемости опухолей гипофиза была определена в аутопсийных и прижизненных КТ и МРТ исследованиях, проведенных по другим показаниям, и варьирует от 10,6 до 16,6%. Инциденталомы гипофиза встречаются примерно с равной частотой среди лиц разного пола и возрастных групп, и подавляющее большинство из них - микроаденомы. Макроинциденталомы обнаруживаются у 0,2% пациентов в ходе КТ головного мозга для уточнения состояния ЦНС и у 0,16% пациентов в ходе МРТ в общепопуляционных когортных исследованиях.
КЛАССИФИКАЦИЯ
См. «Гормонально-неактивные опухоли гипофиза».
КЛИНИЧЕСКАЯ ХАРАКТЕРИСТИКА
Исходя из определения инциденталомы гипофиза (образование гипофиза, не сопровождающееся явными клиническими симптомами нарушения гормональной секреции), следует большее внимание уделить активному расспросу пациента, чтобы выявить возможные скрытые клинические проявления (симптомы повышения гормональной активности).
ЭТИОЛОГИЯ
См. «Гормонально-неактивные опухоли гипофиза».
МЕХАНИЗМ РАЗВИТИЯ
См. «Гормонально-неактивные опухоли гипофиза».
ДИФФЕРЕНЦИАЛЬНО-ДИАГНОСТИЧЕСКИЕ МЕРОПРИЯТИЯ
Диагностический алгоритм
При случайно обнаруженной опухоли гипофиза нарушают обычный алгоритм «клинические проявления → гормональные нарушения → топическая диагностика». Поэтому основные задачи диагностического поиска сводят к выявлению/исключению стертых (субклинических) нарушений гормональной секреции аденогипофиза, а также уточнению неврологических и офтальмологических нарушений, которые могут сопутствовать опухолям гипофиза размерами более 10-15 мм.
Анамнез и физикальное обследование
При размерах опухоли гипофиза менее 10 мм при сборе анамнеза и физикальном обследовании в первую очередь необходимо исключить проявления избытка гормональной продукции, характерные для пролактином, акромегалии и болезни Иценко-Кушинга (см. соответствующие главы). При размерах опухоли более 10-15 мм необходимо также исключить неврологические и зрительные нарушения, а также начальные клинические симптомы гипопитуитаризма (см. «Синдром приобретенного гипопитуитаризма у взрослых»).
Лабораторные исследования
При выявлении инциденталомы гипофиза необходимо провести лабораторное исследование для исключения гиперсекреции опухоли гипофиза, а также гипопитуитаризма (уровень доказательности 1).
Исключение гиперсекреции опухоли гипофиза
-
Малая дексаметазоновая проба и/или определение содержания св. кортизола вечером в слюне и/или определения уровня св. кортизола в суточной моче (скрининг болезни Иценко-Кушинга).
-
При наличии симптомов тиреотоксикоза - определение содержания ТТГ и св. Т4 .
-
При наличии репродуктивных нарушений - определение ЛГ, ФСГ и периферических половых стероидов (эстрадиола у женщин и тестостерона у мужчин).
Исключение гипопитуитаризма (см. «Синдром приобретенного гипопитуитаризма у взрослых»).
Инструментальные исследования
Если инциденталома гипофиза была обнаружена в ходе КТ головного мозга, всем пациентам необходимо выполнять МРТ (если нет противопоказаний) для более детальной визуализации опухоли гипофиза (уровень доказательности 1).
Показания к консультациям специалистов
При распространении опухоли гипофиза за пределы турецкого седла, а также при сдавлении хиазмы по данным МРТ необходимы:
ЛЕЧЕНИЕ
При подтверждении гормональной гиперсекреции опухоли гипофиза - лечение больных согласно соответствующим клиническим рекомендациям (см. соответствующие главы).
При отсутствии гормональной гиперсекреции - лечение больных согласно рекомендациям для гормонально-неактивных опухолей гипофиза (ГНОГ) (см. «Гормонально-неактивные опухоли гипофиза»).
При выявлении гипопитуитаризма - проведение соответствующей заместительной терапии (см. «Синдром приобретенного гипопитуитаризма у взрослых»).
СПИСОК ЛИТЕРАТУРЫ
-
Bancos I., Natt N., Murad M.H., Montori V.M. Evidence-based endocrinology: illustrating its principles in the management of patients with pituitary incidentalomas // Best Pract. Res. Clin. Endocrinol. Metab. - 2012 Feb. - Vol. 26 (1). - P. 9-19.
-
Ezzat S., Asa S.L., Couldwell W.T., Barr C.E., Dodge W.E., Vance M.L., McCutcheon I.E. The prevalence of pituitary adenomas: a systematic review // Cancer. - 2004 Aug 1. - Vol. 101 (3). - P. 613-619.
-
Freda P., Katznelson L., Molitch M. The Hormone Foundation?s Patient guide to pituitary incidentaloma assessment and treatment // J. Clin. Endocrinol. Metab. - 2011 Apr. - Vol. 96 (4). - P. 35A-36A.
-
Freda P.U., Beckers A.M., Katznelson L., Molitch M.E., Montori V.M., Post K.D., Vance M.L.; Endocrine Society. Pituitary incidentaloma: an endocrine society clinical practice guideline // J. Clin. Endocrinol. Metab. - 2011 Apr. - Vol. 96 (4). - P. 894- 904.
-
Molitch M.E. Nonfunctioning pituitary tumors and pituitary incidentalomas // Endocrinol. Metab. Clin. North Am. - 2008. - Vol. 37. - P. 151-171.
-
Fernandez A., Karavitaki N., Wass J.A. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK) // Clin. Endocrinol. (Oxf). - 2010. - Vol. 72. - P. 377-382.
-
Raappana A. 1., Koivukangas J., Ebeling T., Pirilä T. Incidence of pituitary adenomas in Northern Finland in 1992-2007 // J. Clin. Endocrinol. Metab. - 2010. - Vol. 95. - P. 4268-4275.
СИНДРОМ «ПУСТОГО» ТУРЕЦКОГО СЕДЛА
Иловайская И.А.
ОПРЕДЕЛЕНИЕ
Синдром «пустого» турецкого седла - комплекс нейроэндокринных, неврологических и/или нейроофтальмологических нарушений, развивающийся у лиц с пролабированием мозговых оболочек в полость турецкого седла и «распластыванием» гипофиза по дну и стенкам турецкого седла.
Кроме синдрома «пустого» турецкого седла, выделяют еще симптом «пустого» турецкого седла - диагностированное при КТ или МРТ пролабирование мозговых оболочек в полость турецкого седла, повлекшее существенное уменьшение вертикального размера гипофиза (менее 3 мм), но не сопровождающееся какой-либо клинической симптоматикой.
КОД ПО МКБ-10
ЭПИДЕМИОЛОГИЯ
Данные о распространенности синдрома «пустого» турецкого седла различны. По результатам патологоанатомических исследований, «распластанный» уменьшенный гипофиз находят в 5,5-10% случаев.
Наиболее часто синдром «пустого» турецкого седла выявляют в возрасте от 35 до 55 лет, женщины страдают в 5 раз чаще мужчин.
ПРОФИЛАКТИКА
КЛАССИФИКАЦИЯ
По анамнестическому признаку различают первичный и вторичный синдром «пустого» турецкого седла.
-
Первичный синдром возникает без видимых предшествующих заболеваний гипофиза.
-
Вторичный - из-за сокращения размеров или разрушения гиперплазированного гипофиза (или опухоли гипофиза) после операции или облучения, медикаментозного лечения агонистами дофаминовых рецепторов или аналогами соматостатина, после кровоизлияния в опухоль гипофиза или после других состояний.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
При входе в турецкое седло твердая мозговая оболочка расщепляется на два листка, один из которых выстилает стенки и дно турецкого седла, а другой закрывает вход в него и образует диафрагму. В центре диафрагмы имеется отверстие для ножки гипофиза. Прикрепление диафрагмы, ее толщина и размеры отверстия подвержены значительным вариациям. Паутинная оболочка обычно не проникает в полость турецкого седла, хотя под диафрагмой расположена небольшая цистерна гипофиза.
Одно из условий формирования «пустого» турецкого седла - недостаточность диафрагмы. Врожденное недоразвитие диафрагмы турецкого седла, по данным разных авторов, встречается в 40-50% случаев патологоанатомических исследований. Паутинная оболочка пролабирует в полость турецкого седла через отверстие в диафрагме в том случае, если размер его превышает 5 мм.
Недостаточность диафрагмы турецкого седла может быть не только анатомической, но и приобретенной. В период эндокринной перестройки в пубертатном возрасте, при беременности и других физиологических состояниях происходит транзиторная гиперплазия гипофиза и его ножки. Изменения гипофизарных структур можно наблюдать и при отсутствии адекватной терапии по поводу первичной гипофункции периферических эндокринных желез, при длительном приеме контрацептивов и т. п. Избыточное давление на диафрагму, увеличение гипофиза и его ножки с последующей их инволюцией ведет к истончению диафрагмы и увеличению ее отверстия. Повышенное внутричерепное давление (например, при черепно-мозговой травме, при опухолях головного мозга и нейроинфекциях) также увеличивает риск расширения отверстия диафрагмы турецкого седла.
Предрасполагающими состояниями являются:
-
наследственная неполноценность соединительной ткани (наличие «пустого» турецкого седла у родителей и детей);
-
увеличение объема гипофиза и его ножки с последующей инволюцией (лимфоцитарный гипофизит, беременности, кровоизлияние в опухоль или гипофиз);
-
инфекционные заболевания с тяжелым течением (менингит, геморрагическая лихорадка);
-
локальное повышение давления в желудочках мозга при опухолях головного мозга, тромбозе синусов;
-
субарахноидальные кисты, развившиеся в результате оптико-хиазмального арахноидита.
Недостаточность диафрагмы, особенно в условиях повышенного внутричерепного давления, приводит к тому, что мягкие мозговые оболочки распространяются в полость турецкого седла, вызывают уменьшение вертикального размера гипофиза и прижимают его к дну и/или к стенкам седла. Физиологические перепады давления спинномозговой жидкости деформируют гипофиз и могут приводить к постепенному расширению турецкого седла. Формирование «пустого» турецкого седла может также происходить за счет расширения гипофизарной цистерны при уменьшении объема гипофиза вследствие вышеперечисленных причин.
-
Причина эндокринных расстройств при синдроме «пустого» турецкого седла - нарушение гипоталамического контроля над гипофизом в результате изменения нормальной анатомии структур головного мозга, расположенных в хиазмально-селлярной области, а не компрессия клеток гипофиза, которые продолжают функционировать даже при значительной гипоплазии.
-
Причина зрительных нарушений - излишнее натяжение зрительных нервов и их перекреста вследствие нарушения анатомических взаимоотношений (при смещении хиазмы в полость турецкого седла, при смещении ножки гипофиза) или недостаточность кровоснабжения хиазмы.
КЛИНИЧЕСКАЯ КАРТИНА
Отличается вариабельностью и динамичностью - периодическими спонтанными ремиссиями, сменяемостью одного симптома другим.
-
Неврологическая симптоматика встречается у 80-90% пациентов (усугубляется на фоне острой или хронической стрессовой ситуации):
-
головная боль - самый частый симптом (не имеет четкой локализации и изменяется от легкой до выраженной, почти постоянной);
-
вегетативные нарушения (приступы озноба, вариабельность АД, кардиалгии без признаков ишемии миокарда по данным ЭКГ, инспираторная одышка и чувство нехватки воздуха, чувство страха, спастические боли в животе, в конечностях, подъемы температуры тела до субфебрильного уровня без связи с инфекционными и/или вирусными заболеваниями, могут отмечаться синкопальные состояния);
-
-
Эндокринные нарушения (гипоили гиперсекреция различных гормонов гипофиза) варьируют по степени тяжести от субклинических до тяжелых форм:
-
гиперпролактинемия (17-50% случаев), может вызывать расстройства половой функции у мужчин и женщин;
-
частичный или тотальный гипопитуитаризм (20-28% случаев, чаще у мужчин) (см. «Синдром приобретенного гипопитуитаризма у взрослых»);
-
синдромы гиперсекреции гипофизарных гормонов (среди пациентов с болезнью Иценко-Кушинга и акромегалией синдром «пустого» турецкого седла находят в 16 и 10% случаев соответственно);
-
симптомы несахарного диабета (менее 10% случаев) (см. «Несахарный диабет»).
-
метаболические нарушения, ожирение до 50% случаев (см. «Метаболический синдром»).
-
-
В ряде случаев (до 10%) наблюдается сочетание синдрома «пустого» турецкого седла с микроаденомами гипофиза. В таких случаях клиническая картина соответствует гипофизарным нарушениям, связанным с гормональной активностью опухоли гипофиза. Известны случаи сочетания «пустого» турецкого седла с пролактиномами, соматотропиномами, кортикотропиномами.
-
Зрительные нарушения встречаются у 15-57% пациентов, различны по характеру и степени выраженности, подвержены колебаниям (зависят от ликвороциркуляции в арахноидальных пространствах и кровоснабжения хиазмально-зрительного пути):
ДИАГНОСТИКА
Анамнез и физикальное обследование
Четких специфических анамнестических и физикальных признаков синдрома «пустого» турецкого седла нет.
-
Первичный синдром «пустого» турецкого седла можно заподозрить, если были черепно-мозговые травмы, у женщин - длительный прием пероральных контрацептивов, больше трех беременностей.
-
Вторичный синдром «пустого» турецкого седла можно заподозрить после перенесенного нейрохирургического и/или лучевого лечения по поводу опухоли гипофиза, кровоизлияния в гипофиз.
-
КТ и МРТ головного мозга - основной метод диагностики синдрома «пустого» турецкого седла.
Лабораторные исследования
-
Биохимический анализ крови (при наличии метаболических нарушений) - липидный спектр, активность печеночных ферментов.
-
Гормональный анализ крови. При отсутствии жалоб и симптомов, указывающих на эндокринное заболевание, достаточно определить содержание свободного T4 . При выраженной клинической картине (гипопитуитаризм, несахарный диабет и т.д.) проводят более детальное соответствующее обследование.
Инструментальные исследования
Помимо основных изменений в хиазмально-селлярной области, МРТ позволяет выявить косвенные признаки внутричерепной гипертензии, сопутствующие этой патологии, - расширение желудочков и ликворосодержащих пространств.
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Эндокринные, зрительные и неврологические нарушения требуют непременного уточнения состояния хиазмально-селлярной области, поэтому при такой клинической картине необходимо проведение МРТ головного мозга. Это исследование дает достаточно четкую характеристику изменений в полости турецкого седла.
ПОКАЗАНИЯ К КОНСУЛЬТАЦИИ ДРУГИХ СПЕЦИАЛИСТОВ
В зависимости от клинической картины могут потребоваться консультации офтальмолога, невропатолога, гинеколога, андролога.
ПРИМЕРЫ ФОРМУЛИРОВКИ ДИАГНОЗА
Синдром «пустого» турецкого седла. Частичный гипопитуитаризм (вторичный гипотиреоз, вторичный гипогонадизм).
Синдром «пустого» турецкого седла. Ожирение 2 ст.
Синдром «пустого» турецкого седла в исходе нейрохирургического лечения ГНОГ (2001 г.). Пангипопитуитаризм (СТГ-недостаточность, вторичный гипотиреоз, вторичный гипогонадизм, вторичный гипокортицизм). Центральный несахарный диабет.
ЛЕЧЕНИЕ
Цели лечения
Коррекция эндокринных, зрительных, неврологических нарушений.
Показания к госпитализации
Необходимость нейрохирургического лечения.
Немедикаментозное лечение
При избыточной массе тела и метаболических нарушениях - диета, направленная на снижение массы тела.
Медикаментозное лечение
Такое лечение обычно проводят в стационаре под строгим контролем (как временная мера в острой ситуации). Ни одно состояние, сопровождающееся повышенным внутричерепным давлением, не лечат фармакологическими препаратами, лекарственными травами, массажем, иглоукалыванием. Лечение состояний, сопровождающихся повышением внутричерепного давления, направлено на причину заболевания: при опухолях гипофиза проводят их удаление, при нейроин-фекциях - антибактериальную терапию и т.д.
Хирургическое лечение
Показания:
Операция - транссфеноидальная фиксация хиазмы (хиазмопексия), тампонада турецкого седла. Необходимость в подобных операциях возникает крайне редко (менее 2% случаев).
Показания к консультации других специалистов
В зависимости от клинической картины могут потребоваться консультации офтальмолога, невропатолога, гинеколога, андролога, нейрохирурга.
Примерные сроки нетрудоспособности
Определяются индивидуально и зависят от выраженности клинической картины.
Дальнейшее ведение
При наличии эндокринных нарушений - ведение согласно соответствующим рекомендациям.
ПРОГНОЗ
СПИСОК ЛИТЕРАТУРЫ
-
Дедов И.И., Бабарина М.Б., Марова Е.И. Синдром «пустого» турецкого седла: Патогенез, клиника, диагностика, лечение (методическое пособие для врачей). - М.: Литтерра, 2005. - 36 с.
-
Евтушенко С.К., Морозова Т.М., Москаленко М.А. Синдром «пустого» турецкого седла (научный обзор и собственное клиническое наблюдение) // Международный неврологический журнал. - 2010. - №3. - С. 11-17.
-
Клиническая нейроэндокринология / Под ред. И.И. Дедова. - М.: УП-Принт, 2011. - 343 с.
-
Самсонова Л.Н. Офтальмологические нарушения при первично «пустом» турецком седле // Вестник офтальмологии. - 2006. - Т. 122, №4. - С. 41-43.
-
Giustina A., Aimaretti G., Bondanelli M., Buzi F., Cannavò S., Cirillo S., Colao A., De Marinis L., Ferone D., Gasperi M., Grottoli S., Porcelli T., Ghigo E., degli Uberti E. Primary empty sella: Why and when to investigate hypothalamic-pituitary function // J. Endocrinol. Invest. - 2010 May. - Vol. 33 (5). - P. 343-346.
-
Guitelman M., Garcia Basavilbaso N., Vitale M., Chervin A., Katz D., Miragaya K., Herre-ra J., Cornalo D., Servidio M., Boero L., Manavela M., Danilowicz K., Alfieri A., Stalldecker G., Glerean M., Fainstein Day P., Ballarino C., Mallea Gil M.S., Rogozinski A. Primary empty sella (PES): a review of 175 cases // Pituitary. - 2013 Jun. - Vol. 16 (2). - P. 270-274.
СИНДРОМ ГИПОГЛИКЕМИИ
Тлинкина И.В.
ОПРЕДЕЛЕНИЕ
Синдром гипогликемии - клинический симптомокомплекс, развивающийся вследствие нарушений в системе поддержания гомеостаза глюкозы (лабораторно характеризуется снижением концентрации глюкозы в плазме ниже 2,8 ммоль/л при наличии симптомов и ниже 2,2 ммоль/л независимо от наличия симптомов).
ЭТИОЛОГИЯ
См. «Классификация».
МЕХАНИЗМ РАЗВИТИЯ
Повышение клиренса и/или снижение поступления глюкозы в кровь - основа патогенеза синдрома. Глюкоза в физиологических условиях - единственный энергетический субстрат для ЦНС; механизмы, направленные на постоянное поддержание ее концентрации, - гликогенолиз и глюконеогенез, липолиз, протеолиз, торможение утилизации глюкозы инсулинзависимыми тканями.
При снижении концентрации глюкозы в плазме крови происходят следующие изменения:
-
4,9-3,8 ммоль/л - изменение электрической активности головного мозга, снижение секреции инсулина;
-
до 3,8 ммоль/л - повышение секреции контринсулярных гормонов (глюкагон, катехоламины, кортизол, гормон роста);
-
до 3,3 ммоль/л - активация вегетативной нервной системы (вегетативные симптомы);
-
до 2,7 ммоль/л - симптомы дефицита поступления глюкозы в головной мозг (нейрогликопенические симптомы).
При быстром снижении концентрации глюкозы вегетативные и нейрогликопенические симптомы появляются одновременно.
КЛИНИЧЕСКАЯ ХАРАКТЕРИСТИКА
Симптомы гипогликемии неспецифичны, поэтому диагноз не может быть установлен лишь на основании клинической картины.
Диагноз не может быть также установлен на основании только лабораторного обнаружения гипогликемии, так как:
-
без какой-либо клинической симптоматики у здоровых людей после более чем двенадцатичасового голодания и через 2-4 ч после введения глюкозы концентрация ее в плазме может быть ниже 3,3 ммоль/л;
-
лабораторная гипогликемия может быть вызвана поздним или некачественным центрифугированием крови, в этом случае в форменных элементах продолжается гликолиз, что объясняет понижение концентрации глюкозы [для более точного определения концентрации глюкозы в плазме необходимо применять пробирки, в которых содержатся ингибиторы гликолиза (оксалаты, фториды)];
-
определение концентрации глюкозы с помощью глюкометра целесообразно лишь для контроля за лечением СД, так как погрешность прибора - 10-20%, что может привести к получению ложноположительного результата.
Триада Уиппла - «классическая» совокупность симптомов.
-
-
Вегетативные симптомы (связаны с компенсаторной активацией вегетативной нервной системы):
-
Нейрогликопенические симптомы (снижение поступления в ЦНС основного энергетического субстрата - глюкозы): слабость, усталость, снижение внимания, головокружение, зрительные и вербальные нарушения, изменение поведения, судороги, потеря сознания.
-
-
Исчезновение симптомов после приема легкоусвояемых углеводов, внутримышечного/подкожного введения глюкагона или внутривенного введения раствора декстрозы (глюкозы♠ ).
ЭПИДЕМИОЛОГИЯ
Из-за этиологического разнообразия гипогликемического синдрома данные о его распространенности отсутствуют.
КЛАССИФИКАЦИЯ
Клиническая классификация гипогликемического синдрома предусматривает постпрандиальную гипогликемию и гипогликемию натощак.
ДИАГНОСТИКА
Анамнез
При подозрении на синдром гипогликемии необходимо уточнить:
-
наличие у пациента СД (терапия таблетированными сахароснижающими препаратами или инсулином - наиболее вероятная причина гипогликемии у данной группы пациентов);
-
принимает ли пациент ЛС (пентамидин, хинин, салицилаты, сульфаниламиды), способные вызвать гипогликемию;
-
наличие заболеваний с развитием печеночной, почечной или сердечной недостаточности;
Физикальное обследование
-
Гипогликемия - клиническая картина подробно описана выше (см. «Клиническая характеристика»).
-
Гипогликемическая кома: кожные покровы пациента влажные, обычной окраски, тургор мягких тканей нормальный, мышечный тонус нормальный или повышенный, дыхание ровное, неучащенное, АД повышено, пульс учащен, реакция зрачков на свет сохранена.
Лабораторные исследования
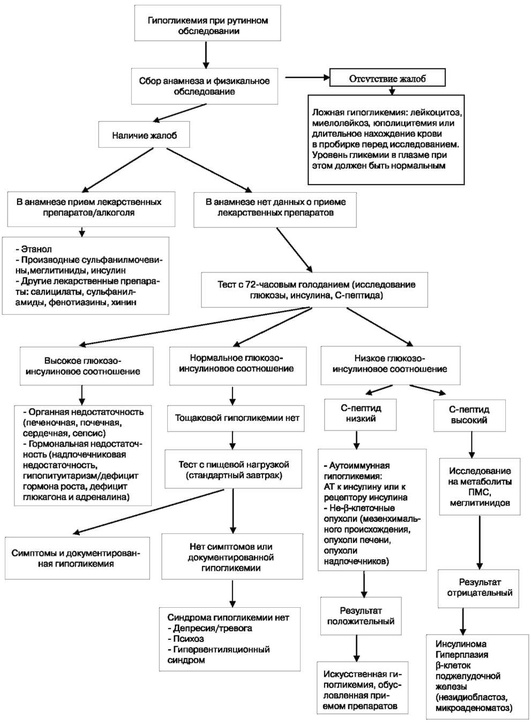
Ниже последовательно изложен диагностический алгоритм гипогликемического синдрома, вытекающий из результатов основных лабораторных показателей.
-
Определение натощак концентрации глюкозы в плазме крови.
-
>3,8 ммоль/л, отсутствие убедительных данных о гипогликемии в анамнезе [симптомы гипогликемии, исчезнувшие после приема углеводов или введения декстрозы (глюкозы♠ )] - исключение гипогликемического синдрома.
-
2,8-3,8 ммоль/л или >3,8 ммоль/л и гипогликемия в анамнезе - проведение пробы с голоданием в течение 72 ч.
-
Проведение пробы требует госпитализации пациента в стационар. Определение гликемии лабораторными методами более достоверно, чем с использованием глюкометра. При проведении пробы допускается прием бескалорийных напитков, не содержащих кофеин. Во время проведения пробы пациент должен сохранять обычную физическую активность. Забор крови (желательно через катетер, поставленный в локтевую вену) производят каждые 6 ч. При концентрации глюкозы ниже 3,4 ммоль/л интервал сокращают до 30-60 мин. Пробу прекращают при концентрации ниже 2,8 ммоль/л и наличии симптомов гипогликемии, которые купируются внутривенным введением раствора декстрозы (глюкозы♠ ).
-
Проба отрицательная (в течение 72 ч у пациента нет снижения концентрации глюкозы <2,8 ммоль/л, отсутствуют клинические проявления) - исключение гипогликемии натощак.
-
Проба положительная (при концентрации <2,8 ммоль/л и развитии клинических проявлений) - переход к следующему этапу диагностического поиска для уточнения этиологии состояния.
-
-
-
Определение C-пептида, иммунореактивного инсулина, метаболитов ПСМ.
-
Низкий уровень C-пептида и иммунореактивного инсулина - гипогликемический синдром, не связан с гиперинсулинизмом, а вызван сопутствующей патологией или приемом ЛС (не сахароснижающих).
-
Низкий уровень C-пептида и высокий иммунореактивного инсулина - введение экзогенного инсулина.
-
Высокий уровень C-пептида и иммунореактивного инсулина, сочетающийся со следующими показателями.
-
-
Достоверные данные о гипогликемических состояниях в анамнезе и отсутствие у пациента гипогликемии натощак - возможна постпрандиальная гипогликемия. В этом случае (особенно при наличии в анамнезе операций на ЖКТ) целесообразно проведение теста с пищевой нагрузкой.
Наличие триады Уиппла после приема пищи - постпрандиальная (или реактивная) гипогликемия. Причинами могут быть нарушение пассажа пищи по ЖКТ после перенесенного оперативного вмешательства или (редко) врожденный дефект ферментов метаболизма углеводов (галактоземия, непереносимость фруктозы).
Отсутствие триады Уиппла - исключение гипогликемического синдрома. Диагноз идиопатической гипогликемии может быть поставлен при отсутствии в анамнезе оперативных вмешательств на ЖКТ и исключении врожденных дефектов метаболизма углеводов.
Инструментальное исследование
Для топической диагностики инсулиномы или гиперплазии инсулярного аппарата:
Показания к консультациям специалистов
При выявлении эндогенного гиперинсулинизма - консультация хирурга-эндокринолога (для проведения топической диагностики, определения объема оперативного вмешательства и его проведения).
Диагностический алгоритм (рис. 14.1)
ЛЕЧЕНИЕ
Тактика лечения зависит от этиологии гипогликемического синдрома.
-
Инсулинома - хирургическое лечение (удаление опухоли путем энуклеации или парциальной резекции поджелудочной железы).
-
Оперативное лечение должен проводить хирург-эндокринолог в условиях специализированного стационара.
-
При невозможности удаления опухоли или при неэффективности оперативного лечения проводится симптоматическая терапия, направленная на профилактику и купирование гипогликемических состояний, а также на предотвращение роста опухоли и ее метастазирования. Для профилактики гипогликемических состояний используют диазоксид в дозе 100-600 мг/сут, которая распределяется на 3-4 приема. При злокачественных инсулиномах применяют аналог соматостатина длительного действия - октреотид, обладающий антипролиферативной активностью. Применяется химиотерапия стрептозотоцином.
-
Для купирования гипогликемических состояний применяют глюкагон в дозе 1 мг подкожно или внутримышечно.
-
-
Гиперплазия инсулярного аппарата поджелудочной железы (незидиобластоз, микроаденоматоз) - хирургическое лечение (резекция 70-80% поджелудочной железы).
-
Оперативное лечение должен проводить хирург-эндокринолог в условиях специализированного стационара.
-
Гипогликемия из-за приема ЛС (не сахароснижающих) - коррекция дозы или отмена препаратов (если назначены не по жизненным показаниям).
-
Тяжелая органная недостаточность (печеночная, почечная, сердечная недостаточность, сепсис), не -β-клеточные опухоли (печени, коры надпочечников), опухоли мезенхимного происхождения:
-
-
дробное питание с достаточным количеством легкоусвояемых углеводов;
Симптоматическое лечение гипогликемии и лечение гипогликемической комы
См. «Гипогликемия и гипогликемический синдром».
Прогноз и дальнейшее ведение пациента
Прогноз и дальнейшее ведение пациента определяются этиологией гипогликемического синдрома.
СПИСОК ЛИТЕРАТУРЫ
-
Гипогликемия: диагностический алгоритм с комментариями // Consilium Medicum. - 2001. - Т. 3, №11. - С. 508-509.
-
Дедов И.И. Клиника и диагностика эндокринных нарушений. - М., 2005.
-
Дедов И.И., Мельниченко Г.А., Фадеев В.В. Эндокринология. - М., 2007.
-
Kar P. Insulinomas may present with normoglycemia after prolonged fasting but glucose-stimulated hypoglycemia // J. Clin. Endocrinol. Metab. - 2006. - Vol. 91, N 12. - P. 4733-4736.
-
Won J.G.S. Clinical features and morphological characterization of 10 patients with noninsulinoma pancreatogenous hypoglycemia syndrome (NIPHS) // Clin. Endocrinol. - 2006. - N 65. - P. 566-578.
-
Williams Textbook of Endocrinology. Tenth edition. Chapter 32. Glucose Homeostasis and Hypoglycemia // Saunders, 2002. - P. 1585-1608.
СИНДРОМ ГИПЕРПРОЛАКТИНЕМИИ
Романцова Т.И.
ОБЩЕПРИНЯТОЕ НАЗВАНИЕ, ОПРЕДЕЛЕНИЕ
Синдром гиперпролактинемии - симптомокомплекс, обусловленный избыточным содержанием пролактина в сыворотке крови, сопровождающийся в большинстве случаев нарушением функции репродуктивной системы у мужчин и женщин. Синдром гиперпролактинемии включает гиперпролактинемический гипогонадизм как самостоятельное нейроэндокринное заболевание (пролактиномы, идиопатическая форма гипогонадизма), а также вторичные (симптоматические) формы гиперпролактинемии, развивающиеся вследствие целого ряда других эндокринных либо соматических заболеваний и состояний.
КОД ПО МКБ-10
ОСНОВНЫЕ ЧЕРТЫ БОЛЕЗНИ
Важнейшей ролью пролактина является регуляция функции репродуктивной системы, соответственно, нарушение секреции гормона прежде всего приводит к нарушению репродукции у женщин и мужчин. В настоящее время открыто множество новых свойств гормона: участие в реализации стрессорного ответа, регуляция функции иммунной системы, контроль нейрогенеза, энергетического обмена, туморогенеза и др., что предопределяет широкий спектр клинических вариантов синдрома гиперпролактинемии. Дофамин - основной фактор, угнетающий секрецию пролактина через систему Д2-рецепторов, локализованных на мембранах лактотрофов. Патогенетическое медикаментозное лечение гиперпролактинемического гипогонадизма основано на ингибирующем эффекте дофамина.
Этиология (причины, классификация) гиперпролактинемии приведена в табл. 14.1.
Физиологические состояния |
Половой акт; физическая нагрузка; лактация; беременность; сон; стресс |
Патологические состояния |
Повреждение гипоталамо-гипофизарной ножки; гранулемы; инфильтративные процессы; облучение; киста кармана Ратке; травма: пересечение гипофизарной ножки, хирургические вмешательства на супраселлярной области; опухоли: краниофарингиома, герминома, метастазы в гипоталамус, менингиома, супраселлярный рост опухолей гипофиза |
Патология гипофиза |
Акромегалия; идиопатическая гиперпролактинемия; лимфоцитарный гипофизит или параселлярные опухоли; макроаденома (компрессионная); плюригормональная аденома; пролактинома; хирургические вмешательства; травма гипофиза |
Системные нарушения |
Грудная клетка - травма грудной клетки, оперативные вмешательства, herpes zoster; ХПН; цирроз печени; облучение головы; эпилептический приступ; СПКЯ; синдром ложной беременности |
Применение фармакологических препаратов |
Анестетики; антиконвульсанты; антидепрессанты; антигистаминные препараты; антигипертензивные препараты; агонисты ацетилхолина; наркотические препараты; стимуляторы высвобождения катехоламинов; блокаторы дофаминовых рецепторов; ингибиторы синтеза дофамина; эстрогены: оральные контрацептивы и их отмена; нейролептики/антипсихотики; нейропептиды; опиаты и антагонисты опиатов |
Макропролактинемия |
|
Эктопическая секреция пролактина |
Дермоидная киста яичников; гипернефрома; бронхогенная карцинома |
Генетические заболевания |
Мутации рецептора пролактина с потерей функции |
Основные клинические проявления синдрома гиперпролактинемии:
Нарушение репродуктивной функции обусловлено гипогонадизмом центрального генеза.
У женщин симптоматика гипогонадизма включает:
Время наступления менархе у многих пациенток несколько запаздывает, в дальнейшем менструации могут быть нерегулярными. Дебют нарушений менструального цикла нередко совпадает с началом половой жизни, отменой пероральных контрацептивов, прерыванием беременности, родами, окончанием грудного вскармливания, введением внутриматочных контрацептивов или оперативным вмешательством.
Бесплодие - одна из основных причин обращения к врачу. При гинекологическом осмотре может отмечаться гипоплазия матки.
Галакторея (выделение молокоподобной жидкости из молочных желез) - первый симптом заболевания лишь у 20% больных, крайне редко - основная жалоба. Степень галактореи колеблется от обильных спонтанных выделений до единичных капель при сильном надавливании на железу. Галакторея может отсутствовать даже при значительном повышении содержания пролактина.
Нередко наблюдаются инволютивные изменения молочных желез и фиброзно-кистозная мастопатия, что обусловлено как эстрогенной недостаточностью, так и дефицитом прогестерона.
Полная клиническая форма заболевания - сочетание аменореи, бесплодия и молокоподобных выделений из молочных желез - преимущественно встречается у пациенток с пролактиномами. При ином генезе отмечают нарушения менструального цикла (чаще по типу опсоменореи) либо неполноценность лютеиновой фазы; обращает на себя внимание обилие жалоб вегетативного характера.
Основные проявления гиперпролактинемии у мужчин:
Гинекомастия и галакторея у мужчин встречаются редко.
При опухолевом генезе заболевания развивается характерная неврологическая симптоматика. Больные жалуются на головные боли постоянного либо транзиторного характера (вследствие давления опухоли на диафрагму турецкого седла, иннервируемую тройничным нервом; распространения опухоли в кавернозные синусы, где проходят I и II ветви тройничного нерва; в результате синусита).
При супраселлярном росте аденомы гипофиза типичны нарушения зрения по типу битемпоральной гемианопсии. Битемпоральные скотомы диагностируют преимущественно при быстро растущих опухолях и переднем положении перекреста зрительных нервов.
Паралич III, IV, V, VI пар черепных нервов возможен при латеральном распространении опухоли и прорастании ее в кавернозные синусы. Клинические проявления этого осложнения: офтальмоплегия, диплопия, птоз. Аденомы с инфраселлярным ростом могут вызывать ликворею и воспалительный процесс в клиновидной пазухе, хотя иногда при таком типе инвазии неврологическая симптоматика отсутствует.
Психоэмоциональные расстройства при гиперпролактинемии:
В ряде случаев развивается специфический психоэндокринный синдром: повышенная раздражительность, тревожность; склонность к депрессивным и психовегетативным реакциям; эмоциональная лабильность; стеничность при выполнении узконаправленной деятельности; пониженная толерантность и аутизм; психосоциальная дезадаптация.
Предположительно, психопатологические нарушения могут быть обусловлены как реакцией пациенток на бесплодие или нарушения менструального цикла, так и дисфункцией нейромедиаторных систем, обусловленной генетически либо перенесенным психическим стрессом.
Среди эндокринно-обменных нарушений заболевания отмечается увеличение массы тела, инсулинорезистентность, хотя распространенность явного СД не превышает таковой в популяции. Увеличение массы тела наблюдается у 60% больных, при этом избыточная масса тела и ожирение I степени встречаются у 1 /3 женщин независимо от формы заболевания, в то время как ожирение II-III степени вдвое чаще развивается при пролактиномах, чем при идиопатической гиперпролактинемии. Возможны изменения липидного состава крови (повышение содержания атерогенных фракций липидов).
При длительной некорригируемой гиперпролактинемии может отмечаться тенденция к снижению МПК.
Механизм развития. Пролактиномы - опухолевые образования моноклональной природы. Они возникают в результате генных нарушений в примордиальной стволовой клетке в период ее дифференцировки. В дальнейшем происходит опухолевая трансформация клетки с последующей клональной экспансией, которая приводит к образованию диагностируемой с помощью инструментальных методов опухоли.
Генез идиопатической формы заболевания по определению не ясен. Не исключено, что основное значение имеют нарушения контроля продукции пролактина на гипоталамическом уровне.
Симптоматические формы обусловлены ослаблением дофаминергического контроля при соответствующих заболеваниях или внегипофизарной продукцией пролактина.
У больных бессимптомной гиперпролактинемией необходимо исключение феномена макропролактинемии. Молекулярная масса обычной молекулы пролактина составляет 23 кДа. Феномен макропролактинемии обусловлен способностью молекул пролактина объединяться либо друг с другом при помощи дисульфидных мостиков (при этом образуется мультимолекулярный комплекс),
либо с белком иммуноглобулином класса G - путем нековалентного связывания. И в том, и в другом случае образуется соединение с молекулярной массой 100-150 кДа. Более медленная элиминация образовавшихся комплексов путем почечного клиренса приводит к увеличению уровня пролактина в сыворотке крови. Высокомолекулярный пролактин проявляет полный спектр биологических свойств in vitro, но не обладает значимой активностью в условиях in vivo.
ЭПИДЕМИОЛОГИЯ
По результатам когортных исследований, распространенность всех форм синдрома гиперпролактинемии варьирует от 0,15 до 1,6% в неселективной взрослой популяции, причем до 80% случаев заболевания приходится на молодых женщин в возрасте 25-40 лет.
ДИАГНОСТИКА
Жалобы, анамнез, клиническая симптоматика, данные физикального обследования изложены в разделе «Основные черты болезни».
Лабораторно-инструментальное обследование включает 4 основных этапа:
Лабораторные исследования
Концентрацию пролактина выражают в нг/мл, в мкЕд/мл (1 нг/мл соответствует 30,3 мкЕд/мл) или в мМЕ/мл (1 мМЕ/мл соответствует 21 нг/мл). У здоровых женщин репродуктивного возраста уровень пролактина в сыворотке не превышает 20 нг/мл (600 мкЕд/мл), у мужчин - 15 нг/мл (450 мкЕд/мл). При оценке показателей гормона в случае небольших отклонений от нормы (до 1000 мкЕд/мл) целесообразно проведение повторных (дву- и троекратных) исследований во избежание ошибочных заключений, поскольку уже сама манипуляция по забору крови как стресс-фактор становится причиной умеренной гиперпролактинемии.
Вероятность опухолевого генеза гиперпролактинемии увеличивается при значениях пролактина более 2000-3000 мкЕд/мл.
У больных с явными клиническими проявлениями заболевания основная форма гормона - пролактин с молекулярной массой 23 кДа. Одной из причин гиперпро-лактинемии может быть феномен макропролактинемии, особенно в случаях несоответствия между выраженностью клинической картины заболевания и содержанием пролактина в крови (например, стойкая бессимптомная гиперпролактинемия). Диагностику феномена макропролактинемии проводят методом гель-фильтрации или фильтрации сыворотки крови с помощью полиэтиленгликоля.
При диагностике необходимо учитывать вероятность эффекта петли высокой концентрации пролактина («high PRL hook effect») - занижение фактического содержания пролактина при его чрезмерных концентрациях в неразбавленных образцах сыворотки крови. Этот феномен обусловлен особенностями кулонов-
ского взаимодействия молекул в дисперсионных растворах. «High PRL hook effect» встречается преимущественно при гигантских пролактиномах (свыше 30-50 мм в диаметре). Пренебрежение вероятностью феномена «high PRL hook effect» у больных с макроаденомами на фоне умеренной гиперпролактинемии (в диапазоне 1500-3000 мкЕд/мл) может привести к неверному заключению о наличии ГНОГ и, соответственно, к выбору ошибочной тактики лечения. При несоответствии больших размеров аденомы гипофиза и умеренного повышения уровня пролакти-на рекомендуется последовательное разведение сыворотки крови для исключения ложных результатов.
Для определения причины гиперпролактинемии функциональные пробы [с метоклопрамидом и протирелином (тиролиберина раствором для инъекций♠)] в настоящее время не используют, так как доказана их низкая информативность.
Инструментальные исследования
Основной метод, позволяющий выявить или исключить опухолевые образования гипоталамо-гипофизарной области (макроили микроаденомы), - МРТ головного мозга. КТ используют в случае невозможности проведения МРТ. При наличии макроаденомы необходимо проводить периметрию. Для уточнения состояния матки и яичников проводят УЗИ малого таза.
Критерий постановки диагноза идиопатической гиперпролактинемии - стойкое повышение содержания пролактина в крови при отсутствии явной патологии гипофиза. Такое заключение можно вынести, только убедившись в отсутствии других возможных причин повышения уровня гормона.
ПОКАЗАНИЯ К КОНСУЛЬТАЦИИ СПЕЦИАЛИСТОВ
При наличии опухоли гипофиза необходимы консультации нейрохирурга и офтальмолога (проведение периметрии). В план обследования женщин входит гинекологический осмотр.
СКРИНИНГ
Определение содержания пролактина проводится всем больным с нарушениями функции репродуктивной системы.
ЛЕЧЕНИЕ
Цели лечения, показания к госпитализации, схемы терапии - см. «Пролактинома».
Выбор принципа лечения зависит от причины синдрома.
При гиперпролактинемическом гипогонадизме метод выбора - медикаментозная терапия агонистами дофамина. Реже при пролактиномах возникает необходимость проведения оперативного вмешательства либо лучевой терапии (более подробно - см. «Пролактинома»).
По отношению к рецепторам дофамина, располагающимся на лактотрофах, выделяют неселективные (бромокриптин) и селективные (хинаголид, каберголин) агонисты дофамина. Хотя возможны спонтанные ремиссии заболевания, в большинстве случаев медикаментозную терапию проводят длительно, иногда - пожизненно. Дофаминомиметики не только помогают достичь нормопролактинемии, но и обладают антимитотическим эффектом.
При ином генезе повышения продукции пролактина проводят патогенетическую терапию основного заболевания.
При феномене макропролактинемии лечение не требуется.
В период определения оптимальной дозы дофаминомиметиков уровень про-лактина измеряют ежемесячно, после достижения нормопролактинемии - 1 раз в 6 мес.
При идиопатической форме гиперпролактинемии, когда существует возможность ремиссии, целесообразно ежегодно отменять препараты (на срок 1-2 мес), контролируя при этом уровень пролактина в крови и отслеживая симптоматику. При опухолевом генезе заболевания плановая отмена препаратов возможна 1 раз в 2 года. При пролактиномах необходимо ежегодно проводить МРТ головного мозга.
В случае наступления беременности агонисты дофамина отменяют. При отсутствии опухоли тактика наблюдения за беременностью и ведение родов обычные, грудное вскармливание разрешено. Вопрос о необходимости возобновления терапии решают после завершения периода кормления.
Критерии эффективности лечения:
СПИСОК ЛИТЕРАТУРЫ
-
Дедов И.И., Мельниченко Г.А., Дзеранова Л.К. и др. Федеральные клинические рекомендации по гиперпролактинемии: клиника, диагностика, дифференциальная диагностика и методы лечения // Пробл. эндокринол. - 2013. - №6. - С. 19-26.
-
Дедов И.И., Мельниченко Г.А., Романцова Т.И. Синдром гиперпролактинемии. - М.: Триада, 2004. - 304 с.
-
Мельниченко Г.А., Марова Е.И., Дзеранова Л.К. и др. Гиперпролактинемия у женщин и мужчин: Пособие для врачей. - М., 2007. - 56 с.
-
Casanueva F.F., Molitch M.E., Schlechte J.A. et al. Gudelines of the Pituitary Society for the diagnosis and management of prolactinomas // Clin. Endocrinol. - 2006. - Vol. 65. - Р. 265-273.
-
Colao A. Pituitary tumours: the prolactinoma // Best Pract. Res. Clin. Endocrinol. Metab. - 2009. - Vol. 23 (5). - P. 575-596.
-
Devi Y., Halperin Y. Reproductive actions of prolactin mediated through short and long receptor isoforms // Mol. Cell Endocrinol. - 2014. - Vol. 382 (1). - P. 400-410.
-
Grattan D.R., Kokay I.C. Prolactin: a pleiotropic neuroendocrine hormone // J. Neuroendocrinol. - 2008. - Vol. 20. - P. 752-763.
-
Jillam M.P., Molitch M.E., Lombardi G. et al. Advances in the treatment of prolactinomas // Endocrine Reviews. - 2006. - Vol. 27 (5). - Р. 485-534.
-
Klibanski A. Prolactinomas // N. Engl. J. Med. - 2010. - Vol. 362 (13). - P. 1219- 1226.
-
Kars M., Dekkers O.M., Pereira A.M., Romijn J.N. Update in prolactinomas // Neth J. Med. - 2010. - Vol. 68 (3). - P. 104-112.
-
Melmed S., Casanueva F.F., Hoffman A.R. et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline // J. Clin. Endocrinol. Metab. - 2011. - Vol. 96 (2). - P. 273-288.
-
Bernard V., Young J., Chanson P., Binart N. New insights in prolactin: pathological implication // Nat. Rev. Endocrinol. - advance online publication 17 March 2015.
СИНДРОМ ПРИОБРЕТЕННОГО ГИПОПИТУИТАРИЗМА (У ВЗРОСЛЫХ)
Иловайская И.А.
СИНОНИМЫ
Гипофизарная недостаточность, гипоталамо-гипофизарная недостаточность.
ОПРЕДЕЛЕНИЕ
Гипопитуитаризм - заболевание, обусловленное частичным или полным нарушением секреции тропных гормонов в аденогипофизе и проявляющееся недостаточностью функции соответствующих органов периферической эндокринной системы.
КОД ПО МКБ-10
Е23.0 Гипопитуитаризм.
ЭПИДЕМИОЛОГИЯ
В основном приобретенный гипопитуитаризм отмечают у женщин в возрасте 30-60 лет, поскольку заболевания, приводящие к развитию гипопитуитаризма, чаще возникают у женщин.
Гипопитуитаризм развивается у 50-90% женщин с послеродовой массивной кровопотерей в анамнезе, у 45-75% пациентов с макроаденомами гипофиза, у 60-70% больных с лимфоцитарным гипофизитом, у 78-90% больных с краниофарингиомами, у 70-80% пациентов с кистами кармана Ратке, у 90-99% больных с супраселлярными дисгерминомами и у 40-60% пациентов с другими новообразованиями ЦНС с локализацией в области зрительного перекреста, у 10-12% пациентов с саркоидозом, у 67-70% пациентов с гистиоцитозом, у 20% больных с туберкулезом ЦНС, у 5-20% пациентов с синдромом «пустого» турецкого седла, у 70-80% больных после тяжелой травмы головного мозга и у 70-90% пациентов после облучения гипоталамо-гипофизарной области.
ПРОФИЛАКТИКА
Профилактика гипопитуитаризма заключается в адекватном своевременном лечении состояний, в результате которых может развиться гипопитуитаризм.
Быстрое восстановление объема циркулирующей крови в случае массивного послеродового кровотечения с целью профилактики послеродового ишемического некроза гипофиза (синдром Шиена).
Лечение гемохроматоза и обследование членов семьи для предотвращения гипопитуитаризма и других осложнений гемохроматоза. Частота гемохроматоза составляет 0,3-0,5%, и его можно легко диагностировать при исследовании концентрации железа и трансферрина в крови. Гипопитуитаризм развивается у небольшого числа больных гемохроматозом, и наиболее часто бывают поражены гипофиз и гонады.
СКРИНИНГ
Скрининг включает исследование функций гипоталамо-гипофизарной системы у лиц с анатомическими нарушениями гипофиза и/или гипоталамуса, а также после воздействий, которые могли бы вызвать повреждение этих функций, т.е. у следующих пациентов.
-
Пациенты с инфильтративными заболеваниями головного мозга или гипофиза.
-
Лица, перенесшие лучевое лечение по поводу негипофизарных новообразований с облучением головы (гипофиза).
-
Больные после нейрохирургического вмешательства и/или лучевого лечения по поводу аденомы или других новообразований гипофиза.
-
Больные с массивной кровопотерей в родах и послеродовой артериальной гипотензией.
КЛАССИФИКАЦИЯ
-
-
Первичный гипопитуитаризм (возникающий вследствие непосредственного разрушения/удаления клеток аденогипофиза).
-
Вторичный гипопитуитаризм (развившийся в результате анатомических и функциональных расстройств гипоталамо-гипофизарных взаимоотношений и ослабления регулирующего влияния гипоталамуса на функции гипофиза).
-
ЭТИОЛОГИЯ
Причины приобретенного гипопитуитаризма у взрослых приведены ниже.
-
Последствия облучения области гипофиза (телегамматерапия и рентгенотерапия, протонотерапия).
-
Прочие (гемосидероз, инфекционные и инфильтративные заболевания и др.).
-
Последствия облучения области гипоталамуса (телегамматерапия и рентгенотерапия).
-
Опухоли ЦНС, локализующиеся в гипоталамической области (астроцитома, краниофарингиома, менингиома и др.).
-
Прочие (поражение гипоталамуса токсического, инфильтративного, инфекционного и иного генеза).
ПАТОГЕНЕЗ
Гипопитуитаризм развивается вследствие повреждения самих гипофизарных клеток после лучевого, аутоиммунного или иного воздействия и/или ослабления гипоталамического контроля над функциями гипофиза. Рилизинг-гормоны контролируют не только функциональную, но и пролиферативную активность клеток аденогипофиза, и при нарушении гипоталамической регуляции постепенно развивается атрофия клеток аденогипофиза.
Патогенетическим фактором развития гипопитуитаризма при наличии опухоли гипофиза служит также повышение давления непосредственно в полости турецкого седла.
Клинические проявления недостаточности тропных гормонов гипофиза возникают, если повреждено не менее 70-75% клеток аденогипофиза. Признаки пангипопитуитаризма развиваются в тех случаях, когда разрушено не менее 90% клеток.
Выпадение функций различных тропных гормонов гипофиза, наблюдающееся в результате хронического патологического процесса (например, при росте макроаденомы гипофиза) или после облучения, происходит в большинстве случаев в определенной последовательности: вначале отмечают нарушения секреции СТГ, затем секреции гонадотропных гормонов (ЛГ и ФСГ), далее ТТГ и в последнюю очередь АКТГ. Однако эта последовательность наблюдается не во всех случаях.
КЛИНИЧЕСКАЯ КАРТИНА
Симптомы гипопитуитаризма могут развиваться на протяжении длительного времени (например, в результате роста гормонально-неактивной макроаденомы гипофиза).
Вначале больных могут беспокоить неспецифические жалобы (быстрая утомляемость, общая слабость, снижение толерантности к физическим нагрузкам), и с момента их появления до установления диагноза может пройти значительный период времени (до нескольких лет).
Клинические проявления гипопитуитаризма многообразны и зависят от выраженности и тяжести недостаточности каждого из гормонов гипофиза.
-
-
Повышение доли жировой ткани в организме из-за увеличения количества висцерального жира и снижения относительного содержания мышечной массы.
-
Уменьшение мышечной силы и выносливости к физическим нагрузкам.
-
Увеличение содержания холестерина с повышением риска развития атеросклероза.
-
Истончение и сухость кожных покровов, уменьшение потоотделения.
-
Психологические нарушения: склонность к апатии, депрессии, неадекватно заниженная самооценка, снижение способности к социальной адаптации.
-
-
Гонадотропная недостаточность (вторичный гипогонадизм).
После нейрохирургического вмешательства по поводу опухоли гипофиза значительных размеров или после обширного кровоизлияния в гипофиз симптомы гипопитуитаризма (выраженная слабость, атония кишечника, падение АД) могут развиваться быстро в течение нескольких часов или суток.
ДИАГНОСТИКА
Диагностические мероприятия при приобретенном гипопитуитаризме проводятся в несколько этапов:
-
1-й этап - предположение о наличии дефицита одного или нескольких гормонов аденогипофиза на основании данных анамнеза и клинических симптомов;
-
2-й этап - соответствующие лабораторные исследования (в зависимости от предполагаемого вида гипофизарной недостаточности);
-
3-й этап - уточнение состояния органов и систем (биохимический анализ крови, остеоденситометрия и другие методы исследования), а также выяснение причины гипопитуитаризма (например, визуализация гипоталамо-гипофизарной области при помощи МРТ).
Анамнез
При сборе анамнеза необходимо уточнить, были ли травмы головного мозга, воздействие ионизирующего излучения на область головы или весь организм, нейрохирургические вмешательства в гипоталамо-гипофизарной области, случаи гемосидероза в семье. У женщин следует уточнить наличие родов с массивной кровопотерей в раннем послеродовом периоде в анамнезе. Эпизод сильной резкой головной боли с выраженным нарушением самочувствия может указывать на перенесенную апоплексию гипофиза. Прогрессирующее снижение остроты зрения и/или сужение полей зрения могут быть признаками новообразования гипофиза.
При сборе жалоб необходимо уточнить общее состояние пациента (слабость, повышенная утомляемость), наличие сонливости, снижения памяти и аппетита, тошноты и рвоты (особенно по утрам или при повышенных нагрузках), непереносимости холодных температур, уменьшения мышечной силы, снижения либидо, бесплодия, у женщин - нарушений менструального цикла, у мужчин - эректильной дисфункции, снижения потенции.
Физикальное обследование
При физикальном обследовании следует обратить внимание на следующие признаки:
Лабораторные исследования
Необходимо проводить исследования, направленные на диагностику недостаточности каждого тропного гормона аденогипофиза.
-
Недостаточность гормона роста.
-
Исследование концентрации гормона роста при стимуляционных пробах (тест с инсулиновой гипогликемией, соматолиберином-аргинином, с клонидином, с глюкагоном) (табл. 14.2).
-
У пациентов с органическим необратимым повреждением гипоталамо-гипофизарной области, дефицитом трех других тропных гормонов и низкими концентрациями ИФР-1 можно сразу диагностировать недостаточность СТГ. Нормальные концентрации ИФР-1 не исключают наличие СТГ недостаточности, но в таком случае необходимо проведение стимуляционного теста.
-
У пациентов с органическим необратимым повреждением гипоталамо-гипофизарной области и дефицитом двух других тропных гормонов для диагностики недостаточности СТГ достаточно провести один стимуляционный тест (инсулинотолерантный тест с инсулиновой гипогликемией).
-
У пациентов с подозрением на наличие только недостаточности СТГ или имеющих дефицит одного другого гипофизарного гормона следует делать 2 стимуляционных теста.
-
По степени повышения концентрации СТГ в ходе тестов можно судить о секреции гормона роста:
-
-
При оценке концентрации ИФР-1 необходимо учитывать, что сопутствующие заболевания печени, прием пероральных эстрогенов, а также плохо контролируемый СД могут приводить к снижению этого показателя.
-
-
Для диагностики недостаточности гонадотропинов проводят исследование базальных концентраций ЛГ и ФСГ, концентрации эстрадиола (у женщин) или тестостерона (у мужчин) в крови (снижение содержания периферических гормонов на фоне сниженных или нормальных концентраций тропных гормонов свидетельствует о нарушении функций гипофиза).
-
Для выявления тиреотропной недостаточности исследуют содержание ТТГ и свободного Т4 в крови - необходимо учитывать, что у части пациентов уровень ТТГ может быть в пределах нормальных референсных значений или даже несколько выше, однако уровень св. Т4 всегда снижен.
-
При подозрении на недостаточность АКТГ показано исследование содержания кортизола в ходе стимуляционных проб (с гипогликемией, вызванной введением инсулина, метирапоном, тетракозактидом) (табл. 14.3). В ходе любого из вышеперечисленных тестов наиболее важным показателем служит пиковая концентрация кортизола, выявляемая в ходе пробы.
| Вводимый препарат | Доза и метод введения | Время забора крови | Ожидаемый пик выброса | Побочные эффекты |
|---|---|---|---|---|
Инсулин |
Внутривенно по 0,1-0,15 Eд/кг массы тела |
За 30 мин до введения, в момент введения и через 15, 30, 45, 60, 90 и 120 мин после введения |
Через 30-60 мин |
Гипогликемия |
Kлoнидин |
Внутрь в дозе 0,15 мг/м2 |
За 30 мин до введения, в момент введения и через 15, 30, 60, 90, 120 и 150 мин после введения |
90-120 мин |
Артериальная гипoтeнзия, сонливость |
Аргинина глyтaмaт (10% раствор) |
Внутривенно медленно по 0,5 мг/кг массы тела, но не более 30 мг |
За 30 мин до введения, в момент введения и через 15, 30, 45, 60, 90 и 120 мин после введения |
30-60 мин |
Покраснение лица, гипергликемия |
Γлюкaгoн |
Внутримышечно по 100 мкг/м2, но не более 1 мг |
В момент введения и через 60, 90, 120, 150 и 180 мин после введения |
120-180 мин |
Тошнота, рвота, поздняя гипогликемия |
Соматолиберинρ |
Внутривенно по 1 мкг/кг массы тела |
За 30 мин до введения, в момент введения и через 15, 30, 45, 60, 90 и 120 мин после введения |
30-60 мин |
| Тест | Условия выполнения теста | Интерпретация результатов |
|---|---|---|
Hизкoдoзиpoвaнный тест с 1-24 АКТГ |
Вводят внутривенно 1-24 АКТГ в дозе 1 мкг. Определение содержания кортизола в крови проводят в момент введения и через 30 и 60 мин после введения. Побочные эффекты не отмечены |
При стимуляции содержание кортизола у здоровых людей должно превышать 750 нмоль/л |
Тест с гипогликемией, вызванной введением инсулина |
Внутривенно вводят инсулин в дозе 0,1-0,15 Eд/кг массы тела до достижения гликемии менее 2,2 ммоль/л. Забор крови осуществляют в момент введения инсулина, через 15, 30, 45, 60, 90 и 120 мин после инфyзии. Побочные эффекты: выраженная гипогликемия, судорожный синдром |
При стимуляции содержание кортизола у здоровых людей после гипогликемии должно превышать 500-550 нмоль/л |
Meтoпиpoнoвый (мeтиpaпoнoвый) тест* |
Meтиpaпoн вводят в дозе 30 мг/кг массы тела в полночь. Концентрацию 11-дeзoкcикopтизoлa, кортизола и АКТГ в крови определяют в 8 ч утра до приема мeтиpaпoнa и в 8 ч следующего утра после приема. Побочные эффекты: тошнота после приема мeтиpaпoнa |
У здоровых лиц содержание 11 -дeзoкcикopтизoлa повышается более 200 нмоль/л, АКТГ - более 150 пг/мл, а концентрация кортизола составляет не более 140 нмоль/л |
* - в настоящее время в РФ не поступает.
Лабораторные исследования, с помощью которых оценивают состояние органов и систем, перечислены ниже.
Инструментальные исследования
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Пангипопитуитаризм следует дифференцировать в первую очередь с неврогенной анорексией, хроническими заболеваниями печени и дистрофической миотонией.
Неврогенная анорексия - состояние, характеризующееся неправильным (неадекватным) отношением к пище и своему внешнему виду и проявляющееся выраженным истощением вплоть до кахексии. На фоне значительного снижения массы тела прекращаются менструации вследствие снижения выработки гонадотропин-рилизинг-гормона, ЛГ и ФСГ, развивается гипоплазия матки и яичников, но при этом сохраняются вторичные половые признаки. Содержание других гипофизарных и периферических гормонов находится в пределах физиологических значений, могут быть повышены базальные концентрации гормона роста и кортизола. Работоспособность и социальная активность не нарушены.
При алкогольном поражении печени, а также при гемохроматозе у мужчин наряду с общим истощением выявляют атрофию тестикул. Однако в большинстве случаев при лабораторных исследованиях удается установить характер основного заболевания и исключить гипопитуитаризм.
Для больных дистрофической миотонией характерны прогрессирующая мышечная слабость, алопеция и катаракта. Гормональных нарушений при данном заболевании не отмечают.
ПОКАЗАНИЯ К КОНСУЛЬТАЦИИ ДРУГИХ СПЕЦИАЛИСТОВ
ПРИМЕР ФОРМУЛИРОВКИ ДИАГНОЗА
-
Пангипопитуитаризм (вторичный гипокортицизм, вторичный гипотиреоз, вторичный гипогонадизм, недостаточность СТГ). Синдром «пустого» турецкого седла после трансназального удаления гормонально неактивной опухоли в 2008 г.
-
Частичный гипопитуитаризм (вторичный гипотиреоз, вторичный гипогонадизм). Посттравматическая энцефалопатия после черепно-мозговой травмы в 2004 г.
ЛЕЧЕНИЕ
Цели лечения
Показания к госпитализации
Немедикаментозное лечение
Всем пациентам с гипопитуитаризмом показаны лечебная физкультура и рациональное питание, обогащенное витаминами и белками. Учитывая, что при недостаточности гормона роста, гипотиреозе и гипогонадизме у больных отмечают нарушения липидного обмена, целесообразно уменьшить содержание животных жиров в рационе питания. Отдельным пациентам необходимо давать рекомендации по дополнительному приему препаратов кальция и определенных физических нагрузок для профилактики и лечения остеопении.
Медикаментозное лечение
По результатам лабораторных исследований определяют показания к проведению медикаментозной терапии.
-
Замещение недостатка кортизола. Применяют препараты натурального гидрокортизона, кортизона ацетата (предшественник гидрокортизона, который метаболизируется в печени) и их полусинтетических производных (преднизолон). Дозу препарата подбирают индивидуально. Главным критерием эффективности лечения считают клиническую картину (отсутствие симптомов недостаточности и/или избытка глюкокортикоидов). В утренние часы (с 8 до 12 ч) обычно принимают 65%, остальное - во второй половине дня (после 16 ч). В дебюте заболевания обычно назначают небольшую дозу глюкокортикоидов утром в один прием (например, гидрокортизон по 5-10 мг или кортизона ацетат по 6,25-12,5 мг утром после завтрака). При необходимости дозу увеличивают и распределяют на 2-3 приема. Исследование содержания кортизола и/или АКТГ на фоне приема препаратов не проводят. Поскольку активация синтеза и секреции альдостерона зависит в основном от активности ренина, а не АКТГ, минералокортикоиды при вторичном гипокортицизме чаще не назначают. В стрессовых ситуациях необходимо увеличивать дозу глюкокортикоидов в 1,5-2 раза или вводить их парентерально.
-
С увеличением длительности заболевания потребность в глюкокортикоидах может увеличиваться. Однако суточная доза принимаемых глюкокортикоидов обычно не должна превышать 20 мг гидрокортизона (или его эквивалента). Противопоказаний для назначения глюкокортикоидов нет.
-
При условии компенсации или исключения гипокортицизма показано замещение недостатка Т4 (противопоказаний для этого нет). Применяют препараты левотироксина натрия, обычно после диагностики заболевания назначают полную заместительную дозу (для молодых пациентов - из расчета 1,6 мкг на кг массы тела). Главный критерий эффективности лечения - нормализация концентрации свободного Т4 в крови. Концентрацию ТТГ при этом не определяют, поскольку она не имеет диагностического значения.
-
Заместительная терапия половыми гормонами у мужчин и женщин показана в большинстве случаев.
-
Принципы лечения гипогонадизма у женщин приведены ниже.
-
Предпочтительно применять аналоги натуральных эстрогенов (эстрадиол) и прогестерона (прогестерон, дидрогестерон). В возрасте до 45 лет доза эстрогенов составляет 1-3 мг/сут, в среднем 2 мг/сут (в расчете на эстрадиол). Предпочтительнее принимать эстрогены и гестагены в секвенциальном (циклическом) режиме. Рекомендуют назначать препараты для 28-дневного приема (не имеющие 7-дневного перерыва). После 45-50 лет доза эстрогенов должна быть равна 1-2 мг/сут (в расчете на эстрадиол), и предпочтительнее монофазный режим приема ЛС. Общую длительность терапии эстроген-гестагенными препаратами определяет лечащий врач (ориентировочно до 55-65 лет). Препараты и схемы лечения для женщин с гипогонадотропным гипогонадизмом приведены в табл. 14.4 и 14.5.
-
Если у женщины отсутствует матка (например, удалена вследствие послеродового массивного кровотечения, что бывает у пациенток с синдромом Шиена), показано назначение препаратов, содержащих только эстрогены. Эстрогены можно принимать как внутрь, так и использовать в виде трансдермального геля. Трансдермальное применение эстрогенов имеет определенные преимущества: исключено прохождение активного действующего вещества через печень, поддерживается физиологическое соотношение эстрадиола к эстрону (2:1), лечение можно проводить при заболеваниях гепатобилиарной системы, нарушениях липидного и углеводного обмена.
-
Противопоказания для назначения половых гормонов у женщин перечислены ниже:
-
-
Лечение гипогонадизма у мужчин приведено в разделе «Синдром гипогонадизма у мужчин». Выделяют следующие противопоказания для назначения половых гормонов у мужчин:
-
Контроль эффективности проводимого лечения половыми гормонами включает следующие показатели:
-
-
Терапия гормоном роста показана некоторым пациентам.
-
Данное лечение назначают после компенсации всех других видов гипофизарной недостаточности. Показаниями для терапии гормоном роста служит наличие клинической симптоматики недостаточности данного гормона и снижение концентрации ИФР-1 в крови. Применяют аналоги человеческого гормона роста. Начальная доза препарата для взрослых составляет 0,03-0,04 МЕ/кг массы тела в неделю (ориентировочно 0,4- 0,5 МЕ/сут). Инъекции выполняют в 20-22 ч с помощью шприц-ручки подкожно в живот или бедро. Поддерживающую дозу подбирают индивидуально. Главный критерий эффективности лечения - соответствие концентрации ИФР-1 (которую определяют через 1 мес после каждого увеличения дозы препарата) физиологическим колебаниям, характерным для пола и возраста больного. При необходимости дозу гормона роста увеличивают ежемесячно на 0,2-0,5 МЕ. Средняя ежедневная доза гормона роста у взрослых приблизительно соответствует 0,125-0,25 МЕ/кг массы тела в неделю (ориентировочно 0,8-2,4 МЕ/сут).
-
Возможно развитие побочных эффектов при лечении гормоном роста: отеки (30-40%), артралгии (10-15%), миалгии (5-10%), локальный дискомфорт в местах инъекций. Побочные эффекты обычно отмечают в течение первых месяцев лечения, и в большинстве случаев они бывают транзиторными и не зависят от дозы препарата. В случае возникновения побочных эффектов дозу следует уменьшить до ранее хорошо переносимой.
-
Пациенты должны быть информированы о том, что уменьшение жировой массы, увеличение мышечной массы и улучшение психологического статуса обычно происходят через 3-6 мес лечения, но могут развиваться и более медленно.
-
Потребность в гормоне роста с возрастом снижается. Доза препарата не должна быть постоянна при длительном лечении, и наиболее тщательный контроль над проводимым лечением необходим у пациентов пожилого возраста.
-
Противопоказания для назначения терапии гормоном роста приведены ниже.
-
| ЛС | Ежедневная доза препарата для имитации первой фазы менструального цикла (14 сут приема) | Ежедневная доза препаратов для имитации 2-й фазы менструального цикла (14 сут приема) |
|---|---|---|
Дидрогестерон + эстрадиол (фемостон 2/10♠) |
2 мг эстрадиола перорально |
2 мг эстрадиола и 10 мг дидрогестерона перорально |
Дидрогестерон + эстрадиол (фемостон 1/10♠) |
1 мг эстрадиола перорально |
1 мг эстрадиола и 10 мг дидрогестерона перорально |
Трансдермальные гели: эстрадиол (дивигель♠, эстрожель♠) |
1,5-3 мг эстрадиола |
1,5-3 мг эстрадиола |
Дидрогестерон (дюфастон♠) |
10-20 мг дидрогестерона |
|
Трансдермальные гели: эстрадиол (дивигель♠, эстрожель♠) |
1,5-3 мг эстрадиола |
1,5-3 мг эстрадиола |
Прогестерон (утрожестан♠) |
200 мг микронизированного прогестерона |
| ЛС | Ежедневные дозы и способы применения |
|---|---|
При наличии матки |
|
Эстрадиол (дивигель♠, эстрожель♠) + прогестерон (утрожестан♠) |
0,5-1,5 мг эстрадиола трансдермально +100 мг микронизированного прогестерона во влагалище |
Дидрогестерон + эстрадиол (фемостон 1/5♠) |
1 мг эстрадиола + 5 мг дидрогестерона перорально |
Эстрадиол (дивигель♠, эстрожель♠) + дидрогестерон (дюфастон♠) |
0,5-1,0-1,5 мг эстрадиола трансдермально + 10 мг дидрогестерона перорально |
При удаленной матке |
|
Эстрадиол (дивигель♠, эстрожель♠) |
0,5-0,75-1,0-1,5 мг эстрадиола трансдермально |
Хирургическое лечение
Хирургическое лечение может потребоваться для устранения причины гипопитуитаризма (например, если причиной заболевания служит объемное образование в области зрительного перекреста и турецкого седла).
Показания к консультации других специалистов
Показана консультация гинеколога у женщин и андролога у мужчин.
Дальнейшее ведение
Мониторинг пациентов с гипопитуитаризмом включает периодические обследования для коррекции доз гормональных препаратов и оценки состояния первичного заболевания, вызвавшего гипопитуитаризм.
Необходимость и кратность обследования определяет в каждом конкретном случае лечащий врач.
-
Клиническое обследование (жалобы, измерение массы тела, оценка АД и ЧСС, состояние кожи и волосяного покрова, состояние зрения, соотношение жировой/мышечной массы) необходимо проводить каждые 6-12 мес. У мужчин дополнительно оценивают наличие гинекомастии и исследуют состояние предстательной железы.
-
Лабораторные исследования включают ежегодное определение концентрации пролактина, свободного Т4 , БСПС и тестостерона (у мужчин), ежемесячное изучение содержания ИФР-1 при подборе дозы гормона роста, а после определения дозы гормона роста - каждые 6-12 мес.
-
У женщин на фоне приема препаратов половых гормонов показаны УЗИ органов малого таза каждые 6-12 мес, в возрасте до 45 лет УЗИ молочных желез каждые 12 мес, а после 45 лет маммография каждые 12-24 мес.
-
В случае развития гипопитуитаризма на фоне первично «пустого» турецкого седла, после облучения гипоталамо-гипофизарной области (по поводу внегипофизарных заболеваний), травмы головного мозга, инфаркта гипофиза, лимфоцитарного гипофизита повторно МРТ головного мозга не проводят.
-
В случае наличия опухоли гипофиза или объемного образования гипоталамо-гипофизарной области иной природы показана МРТ головного мозга один раз в 6-18 мес с целью исключения роста данного образования.
Информация для пациента
Гипопитуитаризм - заболевание, при котором происходит снижение или полное прекращение выработки гормонов гипофизом. Гипофиз - эндокринная железа небольшого размера (не более 1 см3 ), располагается в основании головного мозга, в костной выемке под названием «турецкое седло», и вырабатывает жизненно важные гормоны: гормон роста (у детей обеспечивает процессы линейного роста и формирования костей и мышц, а у взрослых контролирует обмен веществ, в том числе углеводный, жировой, водно-солевой обмен), пролактин (регулирует функцию деторождения и обеспечивает грудное вскармливание), АКТГ (контролирует функцию надпочечников), ТТГ (регулирует функцию щитовидной железы), ФСГ и ЛГ (необходимы для функционирования половых желез).
Основными причинами гипопитуитаризма бывают опухоли гипофиза, сосудистые нарушения (аневризмы, артерииты, кровоизлияния в гипофиз, массивные кровотечения при родах), перенесенные энцефалиты, менингиты, инфильтративные или инфекционные заболевания, травмы головного мозга.
Вследствие патологических процессов гипофиз вырабатывает слишком мало гормонов, что приводит к торможению процессов в организме, зависящих от гормонов: снижению функции щитовидной железы и надпочечников, нарушению половой функции, а также к снижению общего тонуса. Самый ранний признак заболевания - уменьшение полового влечения и потенции у мужчин и нарушение менструального цикла (вплоть до полного прекращения менструаций) у женщин. Кроме того, для гипопитуитаризма характерно выпадение волос в подмышечной впадине и на лобке, бледный, восковидный цвет кожи. Часто беспокоят снижение температуры тела и АД, повышенные сонливость и утомляемость, потеря интереса к окружающему миру и к себе. Нередко пациенты отмечают непереносимость голода, возникновение приступов головокружения натощак. Если заболевание вызвано опухолью гипофиза или окружающих его участков мозга, возможно ухудшение зрения.
С подобными проблемами необходимо обратиться к врачу-эндокринологу. Для диагностики заболевания врач может назначить лабораторные исследования, пробы для выявления нарушения секреции гипофизарных гормонов, МРТ головного мозга, консультации других специалистов.
В случае подтверждения гипопитуитаризма в ходе проведенного обследования врач назначает больным гормональную терапию, которую проводят пожизненно с целью восстановления нормальных концентраций соответствующих гормонов в организме. Если причиной болезни служит опухоль, то необходимы хирургическое лечение и/или лучевая терапия с последующим назначением гормональных препаратов. При правильном лечении удается ликвидировать симптомы заболевания и добиться нормальной работы внутренних органов.
ПРОГНОЗ
Имеются данные об увеличении смертности больных с гипопитуитаризмом примерно в 2 раза по сравнению с общей популяцией. Причинами смерти в основном бывают респираторные, цереброваскулярные и сердечно-сосудистые заболевания. К факторам неблагоприятного прогноза относят пол пациента (среди женщин смертность выше), молодой возраст в дебюте заболевания, отсутствие адекватного лечения, а также причины заболевания (краниофарингиома, радиолучевая терапия в анамнезе).
СПИСОК ЛИТЕРАТУРЫ
-
Стандарт медицинской помощи больным с гипопитуитаризмом // Проблемы стандартизации в здравоохранении. - 2008. - №5. - С. 76-78.
-
Боброва Е.И., Павлова М.Г., Сотников В.М., Пронин В.С., Фадеев В.В. Гипо-питуитаризм после облучения гипоталамо-гипофизарной системы // Клиническая и экспериментальная тиреоидология. - 2013. - Т. 9, №3. - С. 15-20.
-
Вакс В.В., Герасименко О.А., Дзеранова Л.К. Приобретенная недостаточность гормона роста у взрослых: этиология, клинические проявления, диагностика и возможности лечения // Ожирение и метаболизм. - 2011. - №2. - С. 11-17.
-
Иловайская И.А. Этиология приобретенного гипопитуитаризма у взрослых // Доктор. Ру. - 2010. - Т. 2, №7 (57). - С. 15-21.
-
Иловайская И.А., Михайлова Д.С., Зекцер В. Ю., Гончаров Н. П., Мельниченко Г.А. Влияние заместительной гормональной терапии на качество жизни пациенток, страдающих гипогонадотропным гипогонадизмом // Вопросы гинекологии, акушерства и перинатологии. - 2008. - Т. 7, №3. - С. 48-55.
-
Клиническая нейроэндокринология / Под ред. И.И. Дедова. - М.: УП-Принт, 2011. - 343 с.
-
Нагаева Е.В. Метаболический эффект гормона роста при гипопитуитризме у взрослых // Ожирение и метаболизм. - 2010. - №1. - С. 21-27.
-
Charmandari E., Nicolaides N.C., Chrousos G.P. Adrenal insufficiency // Lancet. - 2014 Jun 21. - Vol. 383 (9935). - P. 2152-2167.
-
Erfurth E.M. Update in mortality in GH-treated patients //J. Clin. Endocrinol. Metab. - 2013 Nov. - Vol. 98 (11). - P. 4219-4226.
-
Johannsson G., Falorni A., Skrtic S., Lennernäs H., Quinkler M., Monson J.P., Stewart P.M. Adrenal insufficiency: review of clinical outcomes with current glucocorticoid replacement therapy // Clin. Endocrinol. (Oxf). - 2014 Sep 4. - doi: 10.1111/cen. 12603. [Epub ahead of print]
-
Molitch M.E., Clemmons D.R., Malozowski S., Merriam G.R., Vance M.L.; Endocrine Society. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society clinical practice guideline // J. Clin. Endocrinol. Metab. - 2011 Jun. - Vol. 96 (6). - P. 1587-1609.
-
Koulouri O., Auldin M.A., Agarwal R., Kieffer V., Robertson C., Falconer Smith J., Levy M.J., Howlett T.A. Diagnosis and treatment of hypothyroidism in TSH deficiency compared to primary thyroid disease: pituitary patients are at risk of under-replacement with levothyroxine // Clin. Endocrinol. (Oxf). - 2011 Jun. - Vol. 74 (6). - P. 744-749.
СИНДРОМ НЕАДЕКВАТНОЙ СЕКРЕЦИИ АНТИДИУРЕТИЧЕСКОГО ГОРМОНА
Дзеранова Л.К., Пигарова Е.А.
СИНОНИМЫ
Синдром неадекватной секреции АДГ, синдром неадекватной секреции вазопрессина, синдром Пархона, несахарный антидиабет, гиперпексический синдром.
ОПРЕДЕЛЕНИЕ
Синдром неадекватной секреции АДГ - заболевание, характеризующееся постоянной избыточной секрецией АДГ (вазопрессина), не соответствующей изменению осмоляльности крови, что приводит к чрезмерной задержке воды и развитию гипонатриемии.
ЭПИДЕМИОЛОГИЯ
Не определена.
КЛИНИЧЕСКАЯ ХАРАКТЕРИСТИКА
Увеличение массы тела и образование отеков, обусловленные задержкой воды вследствие олигурии, обычно выражены незначительно. Ведущими проявлениями болезни бывают симптомы нарушения сознания, выраженность которых напрямую зависит от степени гипонатриемии.
Физиологическая норма концентрации натрия в крови - 135-145 ммоль/л.
Степени выраженности гипонатриемии:
Так, при гипонатриемии легкой степени выраженности пациентов беспокоят головная боль, увеличение массы тела, анорексия, тошнота, рвота, депрессия, мышечная слабость, сонливость, спазмы мышц, судороги.
При средней степени тяжести гипонатриемии отмечают спутанность сознания, психозы, дезориентацию, спазмы мышц.
При тяжелой гипонатриемии спазмы мышц могут прогрессировать вплоть до общих судорог, может происходить снижение сухожильных рефлексов вплоть до арефлексии, развиваться псевдобульбарный паралич, кома и смерть.
На выраженность клинической симптоматики оказывает непосредственное влияние не только степень снижения концентрации натрия в крови, но и острота развития гипонатриемии. При быстром (менее 24-48 ч) снижении уровня натрия в крови, как правило, проявляется более яркая неврологическая картина, в то время как при постепенном снижении благодаря адаптационным процессам, направленным на выравнивание трансмембранных осмотических градиентов, клинические симптомы могут не соответствовать по тяжести степени гипонатриемии.
ЭТИОЛОГИЯ
Причины синдрома неадекватной секреции АДГ.
-
Заболевания ЦНС: опухоли, воспалительные заболевания, черепно-мозговая травма, оперативные вмешательства на головном мозге.
-
Болезни легких: туберкулез, аспергиллез, пневмонии, эмпиема, саркоидоз, бронхиальная астма, бронхиолит, абсцесс легкого.
-
Эктопическая секреция АДГ: овсяноклеточный и мелкоклеточный рак легкого, мезотелиома, лимфосаркома, тимома, рак поджелудочной железы, простаты, мочеточника и др.
-
ЛС: карбамазепин, винкристин, трициклические антидепрессанты, ингибиторы моноаминооксидазы, нейролептики, фенотиазины, хлорпропамид, тиазидные диуретики, лизиноприл, циклофосфамид.
-
Активирующая мутация в гене рецептора вазопрессина II типа (нефрогенный вариант синдрома неадекватной секреции АДГ).
МЕХАНИЗМ РАЗВИТИЯ
Повышение секреции АДГ центрального или эктопического генеза, так же как и активирующая мутация рецептора вазопрессина II типа, приводит к его практически постоянной активации, независимо от осмоляльности крови и физиологических потребностей организма. При этом происходит избыточная реабсорбция воды в почках, что приводит к водной интоксикации и гипонатриемии, степень выраженности которых зависит от интенсивности синтеза АДГ и объема потребляемой пациентом жидкости.
ДИФФЕРЕНЦИАЛЬНО-ДИАГНОСТИЧЕСКИЕ МЕРОПРИЯТИЯ
Диагностический алгоритм
Диагностические критерии синдрома неадекватной секреции АДГ следующие.
Анамнез
При опросе больного следует подробно выяснить, какие он принимает ЛС (мочегонные, психотропные, слабительные и др.). Необходимо уточнить, соблюдает ли больной низкосолевую диету, ее эффективность, ограничивает ли прием жидкости.
Физикальное обследование
При осмотре специфичных для заболевания симптомов не обнаруживают.
Лабораторные исследования
Исследуют уровень натрия в крови, осмоляльность крови и мочи и почечную экскрецию натрия, а также исключают гипотиреоз (ТТГ, св. Т4 ) и НН (кортизол крови, свободный кортизол в суточной моче и др.).
Нормоволемическую гипонатриемию при синдроме неадекватной секреции АДГ следует дифференцировать от других видов гипонатриемии: псевдогипонатриемии (гиперпротеинемии, гиперлипидемии), гипонатриемии перераспределения (при гипергликемии), гиповолемической гипонатриемии (потеря жидкости через ЖКТ и ожоговые поверхности, НН), гиперволемической гипонатриемии (застойной сердечной недостаточности, избыточного парентерального введения жидкостей, нефротического синдрома, цирроза печени). Для этого назначают пробу с водной нагрузкой, которую проводят утром натощак. Положение больного при пробе должно быть лежачим или полулежачим для исключения возможного повышения АД. Противопоказаниями к проведению пробы служат нарушение выделительной функции почек, наличие гипонатриемии, выраженные отеки, тяжелое соматическое состояние пациента. В начале пробы определяют осмоляльность мочи и крови, содержание натрия в крови, АД, массу тела, отмечаются отсутствие или выраженность отеков, оценивается общее самочувствие пациента. Далее в течение 15-20 мин больной должен выпить жидкость (негазированную столовую воду) из расчета 20 мл/кг массы тела, но не более 1,5 л, или данный объем жидкости вводят внутривенно в виде 0,9% раствора хлорида натрия. В течение последующих пяти часов каждый час измеряют объем и осмоляльность выделенной мочи, АД, массу тела, отмечают наличие отеков и отклонения в самочувствии пациента. На пятом часу проведения пробы дополнительно исследуют осмоляльность крови и концентрацию натрия в ней.
Критерии синдрома неадекватной секреции АДГ по данным пробы приведены ниже.
-
Отсутствие снижения осмоляльности мочи менее 100 мОсм/кг через 2 ч после введения жидкости.
-
Выделение менее 65% выпитой жидкости через 4 ч после введения жидкости.
-
Выделение менее 80% выпитой жидкости через 5 ч после введения жидкости.
-
Развитие гипонатриемии и снижение осмоляльности крови ниже нормы через 5 ч после введения жидкости.
Инструментальные исследования
При проведении дифференциальной диагностики по показаниям назначают МРТ головного мозга, КТ грудной клетки и брюшной полости, УЗИ внутренних органов для исключения опухолевого генеза неадекватной секреции АДГ, а также динамические тесты состояния функции почек.
ЛЕЧЕНИЕ
Цель лечения - нормализация осмоляльности и концентрации натрия в крови, устранение гипергидратации. Тактика лечения зависит от скорости развития гипонатриемии (острая или хроническая), концентрации натрия в крови и состояния больного.
-
При острой (до 48 ч) выраженной гипонатриемии назначают гипертонический раствор натрия хлорида (3%) в сочетании с фуросемидом. Быстрое повышение содержания натрия в крови до уровня, превышающего 125 ммоль/л, опасно, поскольку возможно развитие повреждения ЦНС (например, центральный понтинный миелинолиз). Рекомендуют повышать концентрацию натрия со скоростью 0,5-1 ммоль/л в час до достижения концентрации 125 ммоль/л. Также рекомендуется после достижения регресса неврологической симптоматики гипонатриемии повышать концентрацию натрия в крови не быстрее 8 ммоль в сутки.
-
Самый эффективный способ лечения легкой хронической гипонатриемии - ограничение потребления жидкости до 600-800 мл/сут. При недостаточности эффекта от ограничения жидкости можно назначить флудрокортизон в дозе 0,1-0,4 мг в 1-2 приема в комбинации с препаратами калия для преодоления калийуретического действия препарата. Возможно также назначение препаратов лития, которые вызывают обратимую или частично обратимую форму нефрогенного несахарного диабета с максимальным развитием эффекта через 2-3 нед, но их применение бывает ограничено высокой частотой побочных эффектов. В настоящее время разработаны и применяются за рубежом, но не зарегистрированы в РФ препараты антагонистов рецептора вазопрессина II типа, характеризующиеся высокой эффективностью при лечении синдрома неадекватной секреции АДГ, но безопасность данных препаратов до сих пор обсуждается, поскольку напрямую зависит от контроля скорости коррекции натрия в крови.
СПИСОК ЛИТЕРАТУРЫ
-
Androgue H.J., Madias N.E. Hyponatr emia // N. Engl. J. Med. - 2000. - Vol. 342. - P. 1581-1589.
-
Nephrogenic Syndrome of Inappropriate Antidiuresis / B.J. Feldman et al. // N. Engl. J. Med. - 2005. - Vol. 352. - P. 1884-1890.
-
Spasovski G., Vanholder R., Allolio B., Annane D., Ball S., Bichet D., Decaux G., Fenske W., Hoorn E.J., Ichai C., Joannidis M., Soupart A., Zietse R., Haller M., van der Veer S., Van Biesen W., Nagler E.; Hyponatraemia Guideline Development Group. Clinical practice guideline on diagnosis and treatment of hyponatraemia // Eur. J. Endocrinol. - 2014 Feb 25. - Vol. 170 (3). - P. G1-47. doi: 10.1530/EJE-13-1020. Print 2014 Mar.
-
Пигарова Е.А. Глава 13. Заболевания нейрогипофиза. Клиническая нейроэндо-кринология / Под ред. И.И. Дедова. - М.: УП Принт, 2011. - С. 239-256.
СИНДРОМ ВЫСОКОРОСЛОСТИ
Потешкин Ю.Е., Колода Д.Е.
СИНОНИМЫ
Гигантизм, избыточная длина тела.
ОПРЕДЕЛЕНИЕ
Высокорослость - длина тела выше 97-го процентиля (+2SD от среднего значения) для данного возраста и пола.
На практике необходимо исключать патологические причины высокорослости, даже если рост человека формально укладывается в верхнюю границу нормы. Именно поэтому дети с ростом между 90-м и 97-м процентилями должны находиться под динамическим наблюдением врачей.
ЭПИДЕМИОЛОГИЯ
Высокорослость наблюдается у 2,5% населения.
КЛИНИЧЕСКАЯ ХАРАКТЕРИСТИКА
Клинические проявления зависят от причины развития высокорослости. Для конституциональной (идиопатической) высокорослости характерен высокий рост родителей и сибсов, высокий окончательный рост, отсутствие стигм дисэмбриогенеза, нормальное интеллектуальное развитие, пропорциональное телосложение, своевременное половое развитие, соответствие костного возраста паспортному, нормальные показатели гормона роста и ИФР-I в крови, уровень гормона роста крови на фоне ОГТТ <1 нг/мл.
Для гипофизарного гигантизма, вызванного соматотропиномой гипофиза, характерны грубые черты лица, потливость, как правило, отставание костного возраста от паспортного, повышение уровня гормона роста и ИФР-I, уровень гормона роста на фоне ОГТТ >1 нг/мл, МРТили КТ-признаки аденомы гипофиза.
Для новорожденных с синдромом Сотоса (церебральный гигантизм) характерно увеличение массы и длины тела, выпуклый лоб, долихоцефалия, макроцефалия, высокое «готическое» нёбо, гипертелоризм с антимонголоидным расположением глазных щелей, прогнатия. У детей с синдромом Сотоса наблюдают задержку умственного развития, нарушение координации движений. Кожа и мягкие ткани несколько утолщены. С возрастом рост тела увеличивается, однако пубертатный период обычно наступает раньше, вследствие этого происходит закрытие эпифизов трубчатых костей. Именно поэтому большинство пациентов с синдромом Сотоса в зрелом возрасте имеют нормальный рост.
Клиническую картину синдрома Беквита-Видемана хорошо отражает другое принятое название синдрома - EMG-синдром (омфалоцеле, макроглоссия, гигантизм - от англ. Exomphalos, Macroglossia, Gigantism). Действительно, новорожденный с синдромом Беквита-Видемана имеет крупные размеры, омфалоцеле, макроглоссию, гиперплазию медуллярного слоя почек, гиперплазию островковых клеток поджелудочной железы, нередко становящуюся причиной развития гипогликемии.
Кроме того, при высоком росте отмечается повышенный риск развития некоторых заболеваний и нарушений: идиопатического сколиоза и других нарушений осанки, варикоцеле, спонтанного пневмоторакса, депрессии, остеопороза.
Характерен вид больных с синдромом Марфана или «марфаноподобными синдромами» (гомоцистеинурия, синдром МЭН-2В, врожденная контрактурная арахнодактилия), в первую очередь вследствие уменьшения верхнего сегмента тела (рост сидя) по отношению к нижнему, превышения размаха рук длины тела и арахнодактилии.
Клиническая картина синдрома МЭН-2В - см. «Синдром МЭН-2».
Клиническая картина синдрома Клайнфельтера - см. «Синдром задержки полового развития».
ЭТИОЛОГИЯ
Основные причины высокорослости следующие.
МЕХАНИЗМ РАЗВИТИЯ
К числу самых важных факторов, определяющих конечный рост человека, относят эффекты гормона роста и половых гормонов. Гормон роста - основной стимулятор линейного роста, половые гормоны определяют сроки прекращения ростового эффекта, вместе с тем они сами обладают способностью ускорять линейный рост.
Причины высокорослости могут быть связаны с одним или другим фактором. Например, гипофизарный гигантизм возникает вследствие гиперсекреции гормона роста, а высокорослость при синдроме Клайнфельтера объясняют дефицитом половых гормонов. Однако в ряде заболеваний патогенез высокорослости не установлен.
Влияние генетических факторов на показатель роста достаточно велико. На сегодняшний день подтверждено участие нескольких генов в развитии конституциональной высокорослости, среди них ген белка V типа, связанного с рецептором ЛПНП, ген рецептора витамина D, ген фибриллина 1, ген белка А2 с высокой электрофоретической подвижностью и др.
Ряд специалистов причиной конституциональной высокорослости считают относительную гиперсекрецию гормона роста или гиперчувствительность тканей к гормону роста. По другим данным, важную роль играют ИФР-II и отношение ИФР/ИФР-связывающий белок.
ДИФФЕРЕНЦИАЛЬНО-ДИАГНОСТИЧЕСКИЕ МЕРОПРИЯТИЯ
Самая частая причина рождения крупных детей с массой и/или длиной тела, превышающей норму для данного срока гестации, - СД у матери. Именно поэтому при избыточной длине тела у новорожденного следует в первую очередь искать СД у матери с помощью измерения уровня глюкозы крови. При отсутствии диабета у матери необходимо исключить врожденные заболевания, в первую очередь синдромы Сотоса и Беквита-Видемана.
Диагностический алгоритм
В детском возрасте диагностику высокорослости проводят по алгоритму, представленному на рис. 14.2.
Анамнез
При сборе анамнеза обязательно выясняют:
Физикальное обследование
Физикальное обследование включает:
Лабораторные и инструментальные исследования
Дополнительные методы исследования (проводят строго по показаниям для уточнения диагноза и решения вопроса о лечении) следующие (табл. 14.6).
| Предполагаемое заболевание | Исследования, позволяющие подтвердить диагноз | Результат |
|---|---|---|
Соматотропинома гипофиза |
Уровень гормона роста на фоне ОГТТ МРТ/КТ гипофиза |
Уровень гормона роста на фоне ОГТТ >1 нг/мл, МРТ-или КТ-признаки аденомы гипофиза |
Синдром Клайнфельтера |
Определение кариотипа |
Выявление дополнительной Х-хромосомы у мужчины |
XYY-синдром |
Определение кариотипа |
Выявление дополнительной Y-хромосомы у мужчины |
Гомоцистинурия |
Анализ мочи на гомоцистеин |
Повышение уровня гомоцистеина в моче |
Диагностика остальных заболеваний, вызывающих высокорослость, основана на характерной клинической картине и результатах молекулярно-генетических исследований.
У взрослых дифференциальную диагностику высокорослости проводят с помощью тех же лабораторных и инструментальных диагностических методов. Следует помнить, что значительная часть заболеваний, вызывающих избыточный рост или скорость роста в детском возрасте, не приводит в итоге к высокому росту. Таким образом, высокий рост у взрослого пациента без сопутствующих заболеваний - как правило, проявление конституциональной высокорослости.
В клинической практике важно не только интерпретировать значение роста в данный момент, но и прогнозировать предполагаемый окончательный рост. Данный показатель позволяет оценить ожидаемую прибавку роста до закрытия эпифизарных зон роста. По методу Tanner и Whitehouse необходимо знать хронологический («паспортный») возраст, костный возраст, рост ребенка в настоящий момент. Далее с использованием специальной таблицы коэффициентов [табл. 14.7 (мальчики) и табл. 14.8 (девочки)] рассчитывается окончательный рост.
Возраст, лет |
Коэффициенты |
||||
для роста, см |
для возраста, лет |
для костного возраста, лет |
константа |
остаточное SD, см |
|
4-7,9 |
1,20 |
-7,3 |
0 |
82 |
4 |
8,0- |
1,22 |
-7,2 |
-0,4 |
82 |
3,6 |
8,5- |
1,23 |
-7,0 |
-0,7 |
82 |
|
9,0- |
1,22 |
-6,8 |
-0,8 |
82 |
|
9,5- |
1,21 |
-6,5 |
-0,8 |
82 |
|
10,0- |
1,20 |
-6,2 |
-1,0 |
83 |
|
10,5- |
1,19 |
-5,9 |
-1,2 |
84 |
|
11,0- |
1,16 |
-5,5 |
-1,6 |
89 |
3,5 |
11,5- |
1,13 |
-5,1 |
-2,0 |
94 |
|
12,0- |
1,08 |
-4,2 |
-2,6 |
98 |
|
12,5- |
1,03 |
-3,4 |
-3,2 |
103 |
|
13,0- |
0,98 |
-2,6 |
-3,8 |
108 |
3,1 |
13,5- |
0,94 |
-1,9 |
-4,4 |
113 |
|
14,0- |
0,90 |
-1,4 |
-4,5 |
114 |
2,9 |
14,5- |
0,87 |
-1,0 |
-4,6 |
114 |
|
15,0- |
0,84 |
-0,8 |
-3,8 |
104 |
2,5 |
15,5- |
0,82 |
-0,6 |
-3,1 |
94 |
|
16,0- |
0,88 |
-0,4 |
-2,4 |
71 |
2,0 |
16,5- |
0,94 |
-0,3 |
-1,8 |
48 |
|
17,0- |
0,96 |
-0,2 |
-1,2 |
34 |
0,8 |
17,5- |
0,98 |
-0,1 |
-0,7 |
19 |
|
Возраст, лет |
Коэффициенты |
||||
для роста, см |
для возраста, лет |
для костного возраста, лет |
константа |
остаточное SD, см |
|
4-5,9 |
0,95 |
-6,5 |
0 |
93 |
3,5 |
6,0- |
0,95 |
-6,0 |
-0,4 |
93 |
3,0 |
6,5- |
0,95 |
-5,5 |
-0,8 |
93 |
|
7,0- |
0,94 |
-5,1 |
-1,0 |
94 |
3,2 |
7,5- |
0,93 |
-4,7 |
-1,1 |
94 |
|
8,0- |
0,92 |
-4,4 |
-1,5 |
95 |
2,9 |
8,5- |
0,92 |
-4,0 |
-1,9 |
96 |
|
9,0- |
0,92 |
-3,8 |
-2,3 |
99 |
2,8 |
9,5- |
0,91 |
-3,6 |
-2,7 |
102 |
|
10,0- |
0,89 |
-3,2 |
-3,2 |
106 |
2,9 |
10,5 |
0,87 |
-2,7 |
-3,6 |
109 |
|
До менархе |
|||||
11,0- |
0,83 |
-2,6 |
-3,6 |
114 |
2,9 |
11,5- |
0,82 |
-2,5 |
-3,6 |
115 |
|
12,0- |
0,83 |
-2,4 |
-3,4 |
111 |
2,7 |
12,5- |
0,83 |
-2,3 |
-3,3 |
108 |
|
13,0- |
0,83 |
-2,0 |
-3,1 |
98 |
2,2 |
13,5- |
0,87 |
-1,8 |
-3,0 |
90 |
|
14,0- |
0,91 |
-1,6 |
-2,8 |
79 |
1,2 |
14,5- |
0,95 |
-1,4 |
-2,5 |
67 |
|
После менархе |
|||||
11,0- |
0,87 |
-2,3 |
-3,3 |
100 |
2,6 |
11,5- |
0,89 |
-1,9 |
-3,3 |
91 |
|
12,0- |
0,91 |
-1,4 |
-3,2 |
82 |
2,1 |
12,5- |
0,93 |
-1,0 |
-2,7 |
67 |
|
13,0- |
0,95 |
-0,9 |
-2,2 |
55 |
1,6 |
13,5- |
0,96 |
-0,9 |
-1,8 |
48 |
|
14,0- |
0,96 |
-0,8 |
-1,4 |
40 |
1,2 |
14,5- |
0,97 |
-0,8 |
-1,3 |
37 |
|
Все |
|||||
15,0- |
0,98 |
-0,6 |
-1,1 |
30 |
0,99 |
15,5- |
0,99 |
-0,4 |
-0,7 |
20 |
|
Метод расчета: исходя из возраста с точностью до 6 мес выбирается строка таблицы, затем рост в сантиметрах умножается на коэффициент из 2-го столбца, возраст и костный возраст умножаются на коэффициенты из третьего столбца соответственно и вычитаются из числа, полученного в самом начале, затем к полученному числу прибавляется значение из столбца «Константа», это и будет расчетный рост. Для каждого возраста также рассчитано среднеквадратичное отклонение расчетного роста, отраженное в столбце 6. Пример расчета: девочка, 12 лет, рост 160 см, костный возраст 14 лет, менархе наступило. Расчет будет выглядеть так: 160×0,91-12×1,4-14×3,2+82=166 см, с учетом погрешности окончательный рост может составить 162-170 см.
Показания к консультациям специалистов
ЛЕЧЕНИЕ
Цели лечения
Лечение конституциональной высокорослости
Решение вопроса о необходимости терапевтического вмешательства при конституциональной высокорослости представляет серьезную проблему. Некоторые авторы считают проведение терапии целесообразным, если предполагаемый окончательный рост более трех стандартных отклонений, т.е. для лиц европейской расы больше 195 см для мальчиков и 180 см для девочек. Кроме того, показанием для назначения фармакотерапии некоторые авторы считают идиопатический сколиоз и психосоциальные проблемы, связанные с высоким ростом.
Раньше считали, что основной вклад в рост тела в детском возрасте принадлежит гормону роста, а ускорение роста в пубертатном периоде и закрытие эпифизарных зон роста обусловлены сочетанным действием гормона роста и половых гормонов (андрогенов у мужчин и эстрогенов у женщин). Однако в настоящее время стало понятно: эстрогены играют важную роль в пубертатном скачке роста и закрытии эпифизарных зон роста как у женщин, так и у мужчин.
Таким образом, для лечения высокорослости можно замедлить допубертатный рост, чтобы своевременный пубертат начался при более низком росте, или сократить допубертатный рост, ускорив наступление пубертата.
Терапия половыми гормонами направлена на ускорение наступления пубертатного периода. При рано начатом лечении вероятность уменьшения окончательного роста возрастает. Соответственно, самый подходящий момент для назначения терапии - препубертатный период. Дополнительная трудность - обращение пациентов за медицинской помощью только в середине пубертатного периода.
Лечение конституциональной высокорослости аналогами соматостатина и антагонистами рецептора гормона роста в широкой клинической практике пока не используют.
Лечение девочек
Эстрогены для лечения конституциональной высокорослости применяют с 1950-х годов, однако лишь в конце 1970-х годов на смену старым препаратам (диэтилстилбестрол и другие) пришел синтетический эстроген этинилэстрадиол, до сих пор успешно применяемый в клинической практике.
Девочкам с конституциональной высокорослостью назначают лечение этинил-эстрадиолом в дозе 0,15-0,3 мг/сут. При необходимости дозу можно увеличить до 0,5 мг/сут, если больной хорошо переносит препарат. При возникновении маточных кровотечений «прорыва» показан циклический прием прогестагенов в комбинации с эстрогенами. Лечение необходимо продолжать вплоть до закрытия эпифизов, иначе возможно продолжение роста после отмены эстрогенов.
Назначают этинилэстрадиол внутрь 0,15-0,5 мг 1 раз в сутки + дидрогестерон внутрь по 10 мг 2 раза в сутки с 16 по 25-й день менструального цикла или норэти-стерон внутрь 5 мг 1 раз в сутки с 16 по 25-й день менструального цикла.
При решении вопроса о необходимости терапии эстрогенами следует учитывать частое возникновение побочных эффектов: тошноты, увеличения веса, отеков, повышения АД.
Другие потенциальные проблемы - тромбоэмболия, гиперплазия и рак эндометрия - практически не возникают на фоне приема девочками эстрогенов.
Существуют данные о повышенном риске бесплодия в зрелом возрасте после лечения высокорослости высокими дозами эстрогенов в пубертатном периоде.
Лечение мальчиков
Лечение мальчиков сложнее. В мужском организме эстрогены тоже способствуют закрытию эпифизов, однако назначение экзогенных эстрогенов мальчикам по понятным причинам не рекомендуют. Андрогены также могут ускорить закрытие эпифизарных зон роста, так как вследствие ароматизации в организме происходит превращение андрогенов в эстрогены. Однако на практике необходимость лечения мальчиков препаратами тестостерона возникает крайне редко.
Тестостерон (смесь эфиров) внутримышечно по 250 мг 1 раз в неделю или по 500 мг 1 раз в 2 недели, 6-12 мес.
Наилучших результатов достигают при раннем начале лечения (костный возраст у девочек - 10 лет, у мальчиков - 12,5 лет) - происходит уменьшение конечного роста до 7 см. Назначение терапии после начала пубертатного периода уже не столь эффективно. Назначение половых гормонов при костном возрасте от 14 лет может привести к увеличению окончательного роста.
Лечение высокорослости при других заболеваниях
СПИСОК ЛИТЕРАТУРЫ
-
Гуркин Ю.А. Гинекология подростков: Руководство для врачей. - СПб.: Фолиант, 2000. - С. 546.
-
Диагностика и лечение эндокринных заболеваний у детей и подростков / Под ред. Н.П. Шабалова. - М.: МЕД-пресс-информ, 2003. - 544 с.
-
Колода Д.Е., Пронин В.С. Высокорослость и гигантизм // Врач. - 2005. - №9. - С. 17-21.
-
Колода Д.Е. Высокорослость // Алгоритмы диагностики и лечения. Эндокринные заболевания / Под ред. Г.А. Мельниченко. - М.: Литтерра, 2008.
-
Колода Д.Е. Диагностика и лечение конституциональной высокорослости // Врач. - 2006. - №11. - С. 52-53.
-
Пронин В.С. Современная стратегия диагностики и лечения соматотропи-ном / В.С. Пронин, Ю.Е. Потешкин, Е.П. Гитель, И.В. Васильева, Е.С. Белышева, М.Е. Морозова, Д.Е. Колода, С.Э. Мошенина, Е.В. Чаплыгина, Е.Л. Соркина, Е.В. Пронин, К.Ю. Жеребчикова, Н.А. Чуброва. - М.: ГЭОТАР-Медиа, 2013.
-
Allen D.B., Rose S.R., Reiter E.O. Normal growth and growth disorders // Principles and Practice of Pediatric Endocrinology / Eds Kappy M.S., Allen D.B., Geffner M.E. - Charles C. Thomas, Publisher Ltd. 2005. - P. 77-216.
-
Cohen P. Hyperpituitarism, tall stature, and overgrowth syndromes // Nelson Textbook of Pediatrics, 18th ed. / Eds. R. M. Kliegman, R.E. Behrman, H.B. Jenson, B.F. Stanton. - Saunders, 2007. - 3200 p.
-
Drop S.L.S., Lamberts S.W.J. Constitutional advanced growth and gigantism // Growth Disorders: Pathogenesis and Treatment / Eds. C.J.H. Kelnar, M.O. Savage, H.F. Stirling, P. Saenger. - Chapman & Hall Medical, 1998. - P. 775-789.
-
Iughetti L., Bergomi A., Bernasconi S. Diagnostic approach and therapy of overgrowth and tall stature in childhood // Minerva Pediatr. - 2003. - Vol. 55, N 6. - P. 563-582.
-
Patel L., Clayton P.E. Normal and disordered growth. // Brook?s Clinical Pediatric Endocrinology, 5th ed. / Eds. Savage M.O., Brook C.G. D., Clayton P.E., Brown R.S. - Blackwell Publishing, 2005. - P. 90-112.
-
Patton M.A. Genetic and dysmorphic syndromes with tall stature // Growth Disorders: Pathogenesis and Treatment / Eds C.J. H. Kelnar, M.O. Savage, H.F. Stirling, P. Saenger. - Chapman & Hall Medical, London, 1998. - P. 323-335.
-
Practical Algorithms in Pediatric Endocrinology, 2nd ed. / Ed. Z. Hochberg. - Karger, 2007. - 112 p.
-
Raine J.E. Tall stature // Practical Endocrinology and Diabetes in Children, 2nd ed. / Eds Raine J.E., Donaldson M.D. C., Gregory J.W. et al. - Blackwell Publishing, 2006. - P. 65-68.
-
Simm P.J., Werther G.A. Child and adolescent growth disorders // Australian Family Physician. - 2005. - Vol. 34, N 9. - P. 732-736.
-
Tanner J.M. Prediction of adult height from height, bone age, and occurrence of menar-che, at ages 4 to 16 with allowance for midparent height. / J.M. Tanner, R.H. Whitehouse, W.A. Marshall, B.S. Carter // Arch. Dis. Child. - 1975. - Vol. 50. - P. 14-26.
СИНДРОМ НИЗКОРОСЛОСТИ
Петеркова В.А.
Распознавание причин низкорослости - не очень простая задача, ибо только врожденных генетических синдромов, сопровождающихся низкорослостью, известно более двух тысяч. Из них есть такие ярко очерченные, как синдром Дауна, что у врача не возникает повода для поиска причин низкорослости. В других же случаях необходимо дополнительное обследование и знание проявлений этих синдромов. Ниже представлена классификация заболеваний, которые могут сопровождаться дефицитом роста.
Заболевания, сопровождающиеся задержкой роста (Ranke M.B., 1999)
Идиопатический дефицит гормона роста.
Дефицит гормона роста органического происхождения (органический).
Другие причины низкорослости.
Дефицит роста определяют по перцентильным таблицам вариантов нормального роста. Перцентильные таблицы стандартов роста и веса основаны на измерении национальной репрезентативной когорты детей и подростков с рождения до конечного роста. Перцентили роста и веса (97 (95) -90-75-50-25-10-3 (5)) на данных таблицах представляют отклонения от медианы (50-й перцентиль). 97-й перцентиль соответствует +2 стандартным отклонениям (SD) от среднего, 3-й перцентиль соответствует -2 SD от среднего.
Для решения вопроса о нормальном или патологическом процессе роста и исключения диагностических ошибок на начальном этапе обследования большое значение имеет анализ кривой роста ребенка с учетом границ его конечного роста, рассчитанного на основании среднего роста родителей.
Для определения границ конечного роста, исходя из среднего роста родителей, используется следующая формула:
Прогнозируемый конечный рост для мальчика= [(рост отца + рост матери + 13 см)/2] ±10 см.
Прогнозируемый конечный рост для девочки= [(рост отца + рост матери - 13 см)/2] ±10 см.
Если экстраполируемый конечный рост ребенка по данным роста в момент осмотра, с учетом костного возраста, находится ниже пределов рассчитанного интервала конечного роста, то следует говорить о патологически низком росте.
Оценка пропорциональности скелета важна в первую очередь для исключения различных форм скелетных дисплазий как причин нанизма. В частности, целесообразно вычислять коэффициент «верхний сегмент/нижний сегмент», объем размаха рук.
Степень оссификации эпифизарных зон роста - важный критерий в диагностике нанизма и прогнозе конечного роста. При первичном дефиците роста задержка костного созревания либо отсутствует, либо слабо выражена (скелетные дисплазии, синдромальные формы нанизма, внутриутробная задержка роста, генетическая низкорослость). Для вторичного дефицита роста, особенно для гипофизарного нанизма, характерно значительное отставание костного возраста от хронологического (более двух лет).
Рентгенологическое исследование черепа проводят с целью визуализации формы и размеров турецкого седла и состояния костей черепа. При гипофизарном нанизме турецкое седло нередко малых размеров. Характерные изменения турецкого седла наблюдают при краниофарингиоме: истончение и порозность стенок, расширение входа, супраселлярные или интраселлярные очаги обызвествления. При повышенном внутричерепном давлении видно усиление пальцевых вдавлений, расхождение черепных швов.
При КТ и МРТ головного мозга у пациентов с идиопатическим гипопитуитаризмом выявляют морфологические и структурные изменения: гипоплазию гипофиза, разрыв или истончение гипофизарной ножки, эктопию нейрогипофиза, синдром «пустого» турецкого седла. Проведение КТ и МРТ головного мозга показано при любом подозрении на внутричерепную патологию (объемный процесс).
Конституциональная задержка роста и пубертата (КЗРП) и семейная низкорослость (или нормальная вариантная низкорослость) укладываются в понятие вариантов нормального роста. Дети от родителей с низким ростом, как правило, низкорослы в той же степени, что и родители, за счет генетически запрограммированных потенций роста. Ребенок от родителей, в анамнезе которых задержка роста и пубертата, с большой степенью вероятности унаследуют данный характер развития.
Дети с КЗРП имеют нормальный рост и вес при рождении, растут нормально до 1-2 лет, затем скорость роста снижается и кривая роста находится несколько ниже 3-го перцентиля и параллельна ему. Костный возраст, как правило, отстает от паспортного и соответствует росту, скорость роста - не менее 4 см в год. Пробы на стимуляцию не выявляют снижения уровня СТГ. Пубертат нормальный, но задержан во времени на сроки отставания костного возраста и наступает при достижении костного созревания у мальчиков 11,5-13,0 лет, у девочек - 10,5-12,0 лет. Сроки достижения конечного роста сдвинуты во времени, конечный рост обычно нормальный без гормональной терапии.
К настоящему времени есть многочисленные данные об эффективном лечении детей с КЗРП рекомбинантным гормоном роста человека. Вместе с тем отсутствуют достоверные данные о более высоком конечном росте детей с КЗРП, получавших лечение гормоном роста, несмотря на то, что за определенный период времени (2-3 года) лечение СТГ существенно ускоряет скорость роста.
У мальчиков с КЗРП старше 12 лет с задержкой костного возраста не менее чем на 2 года от хронологического возможно кратковременное лечение короткими курсами небольших доз анаболических стероидов (нероболил♠ , оксандролон, ретаболил♠ ). При этом необходим строгий контроль за ребенком (не реже 1 раза в 6 мес). При быстром прогрессировании костного созревания лечение прекращают.
Гипофизарная карликовость (соматотропная недостаточность)
Соматотропная недостаточность может быть врожденной и приобретенной.
Врожденная соматотропная недостаточность встречается с частотой 1:10 000 новорожденных, может быть обусловлена пороками развития гипофиза или нарушением генов, ответственных за синтез и секрецию гормона роста. Соматотропная недостаточность может быть изолированной при полной сохранности других тропных гормонов гипофиза и множественной, когда выпадают и другие тропные гормоны гипофиза в разной комбинации, вплоть до пангипопитуитаризма. Проведенные исследования О.Б. Безлепкиной и Е.В. Нагаевой в 2004 г. установили, что изолированный СТГ-дефицит наблюдается в 20% случаев, в 20% - пангипопитуитаризм и в 60% - различные комбинации дефицита тропных гормонов.
Приобретенный СТГ-дефицит чаще всего бывает после хирургических вмешательств или радиологического воздействия в гипоталамо-гипофизарной области, при этом чаще наблюдается множественный дефицит тропных гормонов.
Однократное измерение СТГ в крови для диагностики соматотропной недостаточности не имеет диагностического значения вследствие импульсного характера секреции СТГ и возможности получения крайне низких (нулевых) базальных значений СТГ даже у здоровых детей. В связи с этим используют другие современные методы диагностики, а именно: изучение спонтанной секреции гормона роста в крови, определение пика выброса СТГ на фоне стимуляции, исследование ИФР и их связывающих белков в крови.
Провокационные тесты основаны на способности различных фармакологических препаратов стимулировать секрецию и выброс СТГ соматотрофами. В клинической практике наиболее широко используют пробы с инсулином, клонидином, СТГ-рилизинг-гормоном, аргинином, L-Допа (табл. 14.9).
Тотальную соматотропную недостаточность диагностируют при пике выброса СТГ на фоне стимуляции менее 7 нг/мл, частичный дефицит - при пике выброса СТГ от 7 до 10 нг/мл.
| Препарат | Механизм действия | Доза, метод введения | Время забора проб крови, мин | Пик выброса СТГ, мин | Побочные эффекты |
|---|---|---|---|---|---|
Инсулин |
Активация гипоталамических нейронов, стимулирующих секрецию СТГ-рилизинг гормона |
0,1 Ед/кг, в/в |
0-15-30-45-60-90-120 |
30-60 |
Гипогликемия |
Клонидин |
Нейротрансмиттер, адренергический агонист |
0,15 мг/м2, внутрь |
0-30-60-90-120-150 |
90-120 |
Артериальная гипотензия. Сонливость |
СТГ-рилизинг гормон |
Гипоталамический рилизинг-фактор |
1 мкг/кг, в/в |
0-15-30-45-60-90-120 |
30-60 |
- |
L-Допа |
Нейротрансмиттер, допаминергический агонист. Стимулирует освобождение СТГ-рилизинг гормона |
125 мг (вес <15 кг) 250 мг (вес 15-30 кг) 500 мг (вес >30 кг), внутрь |
0-45-60-120-150 |
45-90 |
Тошнота, рвота, головная боль |
Аргинин (L-аргинина гидрохлорид♠) |
Метаболический стимулятор, аминокислота. Стимулирует освобождение СТГ-рилизинг гормона |
0,5 г/кг, 10% р-р, в 0,9% растворе натрия хлорида, в/в, инфузия в течение 30 мин |
0-15-30-45-60-90-120 |
30-60 |
Гипогликемия, покраснение лица |
Глюкагон |
Относительная гипогликемия |
100 мкг/м2, максимальная доза 1 мг, в/м |
0-60-90- 120-150- 180 |
120-180 |
Тошнота, рвота, поздняя гипогликемия |
Необходимое условие проведения СТГ-стимулирующих проб - эутиреоидное состояние щитовидной железы. В случае гипотиреоза необходим предварительный курс лечения тиреоидными препаратами в течение 3-4 нед. Кроме того, дети с ожирением имеют сниженную реакцию СТГ на стимуляцию.
Для одновременной оценки нескольких гипофизарных функций удобно проводить комбинированные тесты с различными гипоталамическими рилизинг-гормонами, в частности инсулин+ тиролиберин + люлиберин-тест, СТГ-рилизинг гормон + тиролиберин + люлиберин-тест, СТГ-рилизинг гормон + кортико-либерин + люлиберин + тиролиберин-тест. Например, при проведении пробы с СТГ-рилизинг гормон + тиролиберин + люлиберин во внутривенную канюлю последовательно вводят СТГ-рилизинг гормон (1 мкг/кг), тиролиберин (7 мкг/кг, максимально 400 мкг), люлиберин (100 мкг). Наличие базальных низких уровней ТТГ и свободного Т4 в сочетании с отсутствием или пролонгированной реакцией ТТГ на тиролиберин говорит о сопутствующем вторичном гипотиреозе. Отсутствие реакции выброса гонадотропинов на люлиберин (повышение более чем в 2 раза от базального уровня) в сочетании с низкими базальными уровнями половых гормонов указывает на вторичный гипогонадизм.
Наиболее диагностически значимые константы в выявлении дефицита СТГ у детей - ИФР, в частности ИФР-I (соматомедин С).
Для лечения гипофизарного нанизма в качестве ЗГТ используют гормон роста человека.
С 1985 г. для лечения детей с соматотропной недостаточностью используют исключительно генноинженерные препараты гормона роста человека. В настоящее время в России прошли клинические испытания и разрешены к использованию следующие препараты соматропина:
Следует отметить, что в настоящее время все дети РФ с подтвержденным диагнозом дефицита СТГ, врожденным или приобретенным, получают бесплатно лечение рекомбинантным гормоном роста. В последние 5 лет - это преимущественно отечественный препарат растан. Ростстимулирующая эффективность всех препаратов сопоставима, серьезные побочные действия не отмечены.
Рекомендуемая стандартная доза гормона роста при лечении классического дефицита СТГ составляет 0,033 мг/ (кг/сут) подкожно, ежедневно в 20:00-22:00.
Лечение необходимо продолжать до закрытия зон роста или до достижения социально приемлемого роста.
При сочетании гипофизарного нанизма со множественным дефицитом гормонов аденогипофиза проводят соответствующую ЗГТ левотироксином натрия (L-тироксин♠ ), глюкокортикоидами (гидрокортизон), половыми стероидами.
Практически неограниченные возможности создания рекомбинантного гормона роста человека расширили потенциальные показания к его применению как у детей, так и у взрослых, которые теперь не ограничиваются рамками только классического гипофизарного нанизма.
К настоящему времени есть данные об эффективном лечении гормоном роста детей с внутриутробной задержкой роста, семейной низкорослостью, синдромами Тернера, Рассела-Сильвера, Прадера-Вилли, анемией Фанкони, гликогенозами, с состоянием после облучения по поводу лейкемии и опухолей мозга, после трансплантации почки, с ХПН, скелетными дисплазиями.
Синдром Шерешевского-Тернера (дисгенезия гонад)
Частота встречаемости синдрома Шерешевского-Тернера составляет 1:2000- 1:5000 новорожденных девочек. Низкорослость служит наиболее общим клиническим проявлением синдрома и встречается у 100% девочек с кариотипом 45ХО. Низкорослость у пациентов с синдромом Шерешевского-Тернера ассоциирована с SHOX геном. Синдром Шерешевского-Тернера должен быть заподозрен в первую очередь у девочек с необъяснимым отставанием в росте. Учитывая наличие мозаичных форм синдрома Шерешевского-Тернера (45ХО/46ХХ, 45ХО/46Х, i (Хq), 46 Xi (Xq), 45ХО/46Х, rX и другие) с минимальным набором типичных клинических симптомов или даже их отсутствием, всем девочкам с задержкой роста (SDS роста ≤ 2 SDS) необходимо исследование кариотипа на первом этапе диагностического поиска.
Характер роста у девочек с синдромом Шерешевского-Тернера представлен умеренной задержкой внутриутробного развития (средняя длина и масса тела новорожденных примерно на 1 SD ниже нормальных показателей и составляет в среднем 48,3 см и 2800 г соответственно) относительно нормальной скорости роста с рождения до 3 лет жизни, прогрессирующим снижением скорости роста с 3 до 14 лет и далее - низкой скоростью роста в течение нескольких лет, с отсроченным закрытием зон роста. Конечный рост при синдроме Шерешевского-Тернера составляет в среднем 142,0-146,8 см. Рост девочек с синдромом Шерешевского- Тернера должен быть представлен на специальных ростовых кривых.
Помимо задержки роста, клиническая симптоматика включает в себя лимфатический отек кистей и стоп в периоде новорожденности, короткую шею с крыловидными складками различной выраженности, низкий уровень роста волос на шее сзади, птоз век (чаще двусторонний), микрогнатию, эпикант, готическое нёбо, деформацию ушных раковин, бочкообразную грудную клетку с широко расставленными сосками, сколиоз, вальгусную девиацию локтевых суставов, деформацию Маделунга, укорочение и утолщение пальцев, типичную дерматоглифику, дисплазию ногтей, множественные пигментные невусы.
Провокационные тесты на стимуляцию гормона роста проводят только у пациенток с синдромом Шерешевского-Тернера, чья ростовая кривая отклоняется от кривой для данного синдрома. Большинство пациенток в детстве имеют нормальный ответ гормона роста на стимуляцию. Сниженные уровни гормона роста в период пубертата связаны, вероятно, с низким уровнем половых стероидов.
Из сопутствующей патологии при синдроме Шерешевского-Тернера часто встречаются врожденные пороки левых отделов сердца (коарктация аорты), врожденные пороки почек и мочевыводящей системы, НТГ, АИТ, средний отит. Также описаны АГ, алопеция, витилиго, патология ЖКТ (кровотечения вследствие сосудистых мальформаций (телеангиоэктазий), гемангиом, флебэктазий; болезнь Крона).
Половой инфантилизм - одна из наиболее общих черт у девочек с синдромом Шерешевского-Тернера. Свыше 90% пациенток имеют дисгенезию гонад, проявляющуюся клинической симптоматикой первичного гипогонадизма и бесплодием. Гормональный профиль характеризуется выраженным повышением уровней гонадотропинов в сыворотке крови, особенно ФСГ. У 10-20% пациенток степень секреции эстрогенов достаточна для индукции спонтанного пубертата и у 2-5% - для спонтанного менархе и беременности без медицинского вмешательства. Это главным образом касается девочек с мозаичным вариантом кариотипа (45ХО/46ХХ) или структурными нарушениями хромосом, с отсутствием крыловидных складок на шее.
Синдром Нунан (Noonan Syndrome)
Клиническая симптоматика при синдроме Нунан сходна с таковой при синдроме Шерешевского-Тернера, однако эти две нозологии - различные заболевания. При синдроме Нунан не описана патология половых хромосом и очевидный путь наследования - аутосомно-доминантный. Приблизительно у 50% пациентов с синдромом Нунан выявляют гетерозиготные миссенс-мутации гена protein-tyrosine pospatase, nonreceptor-type 11, картированного на хромосоме 12q24 (Tartaglia М. и др., 2001). Поражаются дети обоего пола, что говорит о неправомочности такого названия синдрома Нунан, как «мужской Тернер». Как и при синдроме Шерешевского-Тернера, дети могут иметь крыловидные складки на шее, низкий уровень роста волос на шее сзади, птоз, кубитус вальгус, аномалию ушей.
Врожденная сердечная патология включает преимущественно вовлечение правых отделов сердца (клапаны легочного ствола), а не левых отделов сердца, как при синдроме Шерешевского-Тернера. Характерны микропенис и крипторхизм, пубертат часто задержан или неполный. Задержку умственного развития наблюдают в 25-50% случаев.
Синдром Сильвера-Рассела (Silver-Russell Syndrome)
Для синдрома Сильвера-Рассела, впервые описанного Silver с соавт. (1953) и Russell с соавт. (1954), характерны внутриутробная задержка роста с последующим постнатальным дефицитом роста и ряд типичных дисморфичных черт. Заболевание клинически и генетически гетерогенно. При синдроме Сильвера- Рассела описаны нарушения, вовлекающие хромосомы 7, 8, 15, 17 и 18, однако наиболее вероятные кандидаты - хромосомы 7 и 17. Изменения включают сбалансированные транслокации с точками разрыва в 17q23,3-q25, гомозиготную делецию гена ХГЧ на 17q24,1, материнскую юнипатентную дисомию для хромосомы 7 (Hitchins М.Р. и др., 2001). Клиническая симптоматика включает низкую массу и длину тела при рождении, постнатальное отставание в росте. Треугольное лицо маленьких размеров, узкие губы с опущенными уголками («рыбий рот»), укорочение и искривление (клинодактилия) V пальца кистей и стоп, врожденная гемигипертрофия, ведущая к асимметрии туловища и конечностей и нарушению походки, преждевременный пубертат у ряда пациентов.
Внутриутробная задержка роста
Внутриутробная задержка роста (intrauterine growth retardation), или small for gestational age («малые для гестационного возраста»), - клиническое состояние, характеризующееся недостаточным ростом плода. Дефиниция данной патологии подразумевает длину тела при рождении ниже 2 SD для гестационного возраста и пола и/или массу тела при рождении <2500 г при сроке беременности ≥37 нед.
Нарушенный внутриутробный рост обусловлен рядом фетальных и плацентарных факторов. Около одной трети случаев внутриутробной задержки роста вызываются генетическими изменениями, остальные 70% - следствие материнских и/ или плацентарных факторов.
Фетальные факторы включают генетические нарушения, врожденные мальформации, метаболические нарушения, инфекции и многоплодную беременность. Хромосомные аберрации встречаются у 5-7% детей с внутриутробной задержкой роста, из них трисомия 21, трисомия 18 и синдром Тернера - наиболее частые. Недостаточность плаценты - одна из наиболее общих причин внутриутробной задержки роста. АГ, СД, системная красная волчанка, патология почек, коллагеновые сосудистые заболевания - наиболее выраженные факторы риска для развития внутриутробной задержки роста. К факторам риска относят также инфекции (вирусная, бактериальная, протозойная), курение и прием алкоголя во время беременности.
Внутриутробная задержка роста встречается у 2,5-10,0% новорожденных, из них у 8-20% отсутствует постнатальный скачок роста в первые 2 года жизни. Дети с внутриутробной задержкой роста имеют повышенный риск (около 30%) низкорослости во взрослом состоянии. Взрослые пациенты с внутриутробной задержкой роста имеют более высокий риск АГ, ИБС, инсультов, метаболического синдрома (инсулинорезистентность, СД 2-го типа, АГ, гиперлипидемия), чем в общей популяции.
В большинстве случаев низкорослые дети с внутриутробной задержкой роста имеют нормальную секрецию гормона роста. Предполагают, что низкорослость при внутриутробной задержке роста - результат сниженной чувствительности к гормону роста. Однако у части детей с внутриутробной задержкой роста снижена суточная секреция гормона роста и нарушен ее ритм, снижены уровни ИФР-1 и ИФР-связывающего белка-3.
Синдром Прадера-Вилли
Синдром Прадера-Вилли - редкое генетическое заболевание, частота его составляет около 1:16 000 новорожденных. Синдром Прадера-Вилли представляет наиболее частую причину выраженного синдромального ожирения у детей. Молекулярной основой синдрома Прадера-Вилли служит потеря генов короткого плеча отцовской хромосомы 15, как следствие: делеции генов, материнской юнипатентной дисомии, ошибок импритинга (Whittington J.E. и др., 2001).
В периоде новорожденности и раннего детства присутствуют мышечная гипотония, затруднение питания, низкий вес, у мальчиков - микропенис и крипторхизм. В возрасте 2-4 лет развивается ожирение вследствие повышенного аппетита и неконтролируемого приема пищи. Характерны задержка роста, маленькие размеры стоп и кистей, отставание в психомоторном развитии. Умеренная внутриутробная и постнатальная задержка роста сопровождается отсутствием пубертатного скачка роста и низкорослостью во взрослом состоянии: рост мужчин составляет 152-162 см, рост женщин - 145-150 см. Характерны миндалевидные глаза, косоглазие, треугольный рот. Психиатрические и поведенческие проблемы включают навязчивые страхи, депрессию, психозы. У взрослых пациентов с синдромом Прадера-Вилли отмечается повышенная частота дыхательных расстройств во время сна. Во избежание фатальных случаев рекомендуется тщательно контролировать функцию дыхательной системы у всех новорожденных с синдромом Прадера-Вилли и подозревать его у всех новорожденных с мышечной гипотонией.
Основным доказанным нарушением на уровне гипоталамуса служит гонадотропная недостаточность. Вместе с тем все больше данных указывает на то, что при синдроме Прадера-Вилли есть дефицит гормона роста вследствие гипоталамической дисрегуляции, приводящий не только к нарушенному росту, но и к ожирению и дефициту общей массы тела. Ответ гормона роста на стимуляцию инсулином, аргинином глутаматом, клонидином и L-Допа находится на нижней границе нормы или снижен. Аналогичным образом снижена 24-часовая интегрированная концентрация гормона роста и секреция гормона роста, индуцированная сном. Уровни ИФР-1 в крови как снижены, так и нормальные. Есть данные о более высоких уровнях грелина в крови у детей с синдромом Прадера-Вилли, чем у детей с ожирением без синдрома Прадера-Вилли (Butler M.G. и др., 2004).
Синдром Дауна (трисомия 21)
Синдром Дауна - одно из наиболее частых генетических заболеваний, ассоциированных с низкорослостью. В среднем длина тела новорожденного на 2-3 см, а масса тела на 500 г ниже, чем в нормальной популяции. Отставание в росте ребенка сохраняется в постнатальном периоде и сопровождается задержкой скелетного созревания. Пубертатный скачок слабый и также задержан во времени. Конечный рост составляет 135-170 см у мужчин и 127-158 см у женщин. Частота гипотиреоза у пациентов с синдромом Дауна выше, чем в популяции.
Патология костной системы
Врожденная патология костной системы - одна из распространенных причин низкорослости у детей.
Остеохондродисплазии представляют гетерогенную группу заболеваний, характеризующихся патологией непосредственно хряща или кости или того и другого. В настоящее время описано более 100 остеохондродисплазий, классификация которых включает:
-
патологию длинных трубчатых костей и позвоночника, проявляющуюся с рождения (танатоформная дисплазия, ахондроплазия, хондроэктодермальная дисплазия, мезамиелические дисплазии) или в более позднем возрасте (гипохондроплазия, дисхондростеоз, метафизарные хондродисплазии);
-
дефекты развития хряща и фиброзного компонента скелета (множественные хрящевые экзостозы, фиброзная дисплазия);
-
дефекты толщины кортикального слоя диафизов и/или моделирование диафизов (несовершенный остеогенез, ювенильный идиопатический остеопороз, диафизарная дисплазия).
Несмотря на значительный прогресс в выявлении молекулярной основы многих заболеваний, относящихся к группе остеохондродисплазий, клинические и рентгенологические данные остаются ключевыми в диагностике. В большинстве случаев клиническая симптоматика настолько очевидна, что диагноз может быть поставлен уже при рождении или даже пренатально, с помощью УЗИ. В диагностике важен семейный анамнез, хотя многие случаи представляют мутации de novo. Необходимо тщательное измерение пропорций тела, таких как рост сидя, коэффициент «верхний сегмент/нижний сегмент», размах рук, окружность головы. Наиболее частые формы остеохондродисплазий - ахондроплазия и гипохондроплазия.
Ахондроплазия
Ахондроплазия - аутосомно-доминантное заболевание, одна из наиболее частых форм остеохондродисплазий, ее частота составляет 1:26 000 новорожденных. Молекулярной основой ахондроплазии служит мутация в гене рецептора III фактора роста фибробластов (FGFR3), картированного на коротком плече хромосомы 4 (4p16,3) (Naski M.C. и др., 1996). У более 98% пациентов с ахондроплазией выявлена общая мутация в трансмембранном домене FGFR3.
Клиническая картина характерна уже при рождении: крупная голова с большой мозговой частью и резко увеличенными родничками, диспропорциональное телосложение за счет ризомелического укорочения конечностей, короткие широкие кисти и стопы. У детей первого года жизни отмечается разболтанность в коленных суставах. В дальнейшем характерна выраженная задержка роста за счет укорочения конечностей; крупная голова, нависающий лоб, седловидный нос, прогнатизм; укорочение и утолщение пальцев (кисть в виде трезубца), изодактилия; поясничный лордоз (размеры таза уменьшены, таз наклонен вперед); походка раскачивания при ходьбе. Половое развитие не нарушено. Интеллект сохранен. Конечный рост составляет у мужчин около 131 см, у женщин - около 124 см.
Характерный рентгенологический признак - сужение расстояния между корнями дужек поясничных позвонков в каудальном направлении. Отмечается также укорочение диафизов длинных трубчатых костей, укорочение и утолщение фаланг пальцев пястных костей, утолщение и чашеобразное расширение метафизов, деформация суставных поверхностей кистей, относительное удлинение малоберцовой кости. Крылья подвздошной кости укорочены и развернуты, крыши вертлужных впадин горизонтальны. Уменьшение размеров большого затылочного отверстия может вести к развитию гидроцефалии и компрессии спинного мозга в шейном отделе. Сужение спинномозгового канала может быть причиной компрессии спинного мозга, повреждения дисков и, соответственно, неврологической симптоматики в зрелом возрасте.
Гипохондроплазия
Гипохондроплазия - аутосомно-доминантное заболевание, высока частота спорадических случаев. Молекулярные основы гипохондроплазии составляют мутации FGFR3 гена, однако локализация мутаций иная, чем при ахондроплазии, а именно в проксимальном домене тирозинкиназы (TK1) FGFR3-N540K (Asn540Lys) и Ile538Val (Rousseau F. и др., 1995; Grigelioniene G. и др., 1997; Fofanova O. и др., 1998).
Гипохондроплазия характеризуется низким ростом, диспропорционально короткими конечностями ризомелического типа и поясничным лордозом. Характерные рентгенологические изменения: отсутствие увеличения расстояния между отростками позвонков в поясничном отделе позвоночника L1 -L5 или уменьшение этого расстояния, уменьшение объема крыльев подвздошной кости, укорочение длинных трубчатых костей ризомелического типа, удлинение малоберцовой кости, неровность контуров метафизов трубчатых костей.
В большинстве случаев родители, страдающие гипохондроплазией, не подозревают о наличии у себя данной патологии. Задержка роста становится заметной у ребенка после 3-4-летнего возраста и постепенно прогрессирует по мере взросления. Диспропорциональность скелета нередко проявляется только в пубертатном возрасте. Пубертатный скачок роста либо отсутствует, либо выражен слабо. Пубертат не нарушен, интеллект сохранен. Конечный рост составляет 130-150 см.
Несовершенный остеогенез
Несовершенный остеогенез (osteogenesis imperfecta) относится к группе диа-физарных дисплазий. Характерно аутосомно-доминантное наследование с высокой пенетрантностью, значительна доля спорадических случаев. Молекулярной основой заболевания служат делеции, вставки или точковые мутации в генах, кодирующих синтез коллагеновых цепей типа I-pro-α (I) и pro-α2 (I), ведущие к нарушению синтеза коллагена.
Клиническая симптоматика включает частые переломы, серо-синие склеры и тугоухость. Зубы прорезываются поздно, характерен кариес. Основной рентгенологический признак - распространенный остеопороз всего скелета. Кортикальный слой истончен на всем протяжении костей. Поперечный размер диафизов костей уменьшен, кости истончены. Классификация согласно генетическим вариантам включает следующие типы:
-
тип 1 (взрослый, умеренная тяжесть костных изменений), аутосомно-доминантный, переломы при рождении или в течение первого десятилетия жизни. Конечный рост ниже, чем у здоровых родственников. Характерны голубые склеры, сколиоз, слабость в суставах, ранняя тугоухость с нормальным (тип 1А) или нарушенным (тип 1В) дентиногенезом;
-
тип 2 (перинатально-летальная форма), аутосомно-доминантный или рецессивный, характеризуется снижением минерализации скелета и компрессионными переломами in utero;
-
тип 3 (подростковый), аутосомно-доминантный или рецессивный, характеризуется тяжелыми прогрессирующими деформациями длинных трубчатых костей вследствие частых переломов, кифосколиозом и низкорослостью. Также встречаются голубые склеры, исчезающие с возрастом, и нарушенный дентиногенез;
-
тип 4, доминантно наследуемый, с широкой вариабельностью клинической симптоматики.
Патология гена SHOX
Гомеобокс, содержащий ген низкорослости (short stature homeobox-containing gene), был изолирован в 1997 г. Ген низкорослости картирован в псевдоаутосом-ном регионе (PAR1) X (Xp22) и Y (Yp11.3) хромосом. Для нормального фенотипа необходимы две копии данного гена.
Патология гена низкорослости ассоциирована с низкорослостью у пациентов с синдромом Шерешевского-Тернера, Лери-Вейла, нанизмом Лангера, идиопатической низкорослостью (Stuppia L. и др., 2003; Binder G. и др., 2004). Изучение значительной по объему (около 1000 пациентов) популяции с идиопатической низкорослостью без явных скелетных аномалий предполагает наличие мутаций гена низкорослости с частотой 1:2000. Считается, что мутации гена низкорослости ответственны за 1/3 потерю роста (около 13 см) при синдроме Шерешевского- Тернера (Rappold G. и др., 2001).
Делеции и, реже, точковые мутации гена низкорослости объясняют мезамиелическую низкорослость, наблюдаемую при синдроме Лери-Вейла (Leri-Weill), и ответственны за вальгусную деформацию и короткие пястные кости. Частота полной делеции гена низкорослости при дисхондростеозе Лери-Вейла составляет 60-80%, частота точковых мутаций гена - 19% (Zinn A., 2001). Дисхондростеоз Лери-Вейла - доминантно наследуемая костная дисплазия, характеризующаяся деформацией Маделунга, мезомелией и низкорослостью. Генетической основой дисхондростеоза Лери-Вейла служит гаплонедостаточность гена низкорослости. Деформация Маделунга рентгенологически проявляется латеральным и дорзальным искривлением укороченной лучевой кости, подвывихом лучезапястного сустава, треугольной формой лучевого эпифиза, преждевременным сращением эпифизов локтевой кости и защемлением костей запястья в патологически сформированном суставе между локтевой и лучевой костью.
Вторичная задержка роста
НАРУШЕНИЕ ПИТАНИЯ
Причиной задержки роста может быть неадекватное питание ребенка, как в количестве, так и в качестве. Нарушенный рост при этих состояниях (например, при квашиоркоре) часто характеризуется повышенными концентрациями базального или стимулированного гормона роста при сниженных концентрациях ИФР-1. Таким образом, данные состояния можно рассматривать как форму нечувствительности к гормону роста.
Неадекватное питание может также осложнять многие хронические заболевания. Анорексия - типичная черта почечной недостаточности и воспалительных заболеваний кишечника, встречается при врожденных («синих») пороках сердца, застойной сердечной недостаточности, заболеваниях ЦНС и других состояниях. Недостаточное поступление пищи наблюдают при соблюдении разнообразных диет, в основном у девочек в период пубертата. Нервная анорексия является крайним вариантом такого добровольного ограничения пищи и при ее наличии до закрытия зон роста часто ведет к нарушенным процессам роста и задержке пубертата.
НАРУШЕНИЕ ВСАСЫВАНИЯ
Заболевания кишечника, связанные с нарушенной абсорбцией белка, могут вызывать задержку роста у детей. Такие нозологии, как болезнь Крона и целиакия, необходимо учитывать в дифференциальной диагностике при необъяснимой низкорослости у ребенка. При болезни Крона в патогенез низкорослости вовлечены не только мальабсорбция, но и хроническое воспаление, анорексия, неадекватное поступление минералов, применение глюкокортикоидов.
ЗАБОЛЕВАНИЯ СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ
Врожденные («синие») пороки сердца и застойная сердечная недостаточность могут быть причинами низкорослости у ребенка. Поскольку сердечные пороки у детей, как правило, врожденные, многие новорожденные имеют сопутствующие признаки дисморфии и внутриутробной задержки роста. Постнатальная задержка роста связана в основном с гипоксией и повышенной потребностью сердца в энергии. Хирургическое вмешательство часто восстанавливает нормальный процесс роста.
ПАТОЛОГИЯ ПОЧЕК
Заболевания почек, сопровождающиеся нарушением функции, могут приводить к выраженной задержке роста. Как уремия, так и почечный тубулярный ацидоз могут проявляться низкорослостью до появления других клинических симптомов. Факторы, вовлеченные в патогенез низкорослости, включают потерю белка, неадекватное образование 1,25-дигидроксихолекальциферола, резистентность к инсулину, хроническую анемию, метаболический ацидоз. Уровни ИФР-1 и ИФР-2, как правило, находятся в пределах нормы, но повышение уровней ИФР-связывающих белков, особенно ИФР-связывающего белка-1, может приводить к подавлению эффектов ИФР. ХПН ассоциирована с рядом изменений в системе «гормон роста-ИФР-ИФР-связывающий белок», а именно с повышенным пульсирующим освобождением гормона роста. При ХПН биоактивность ИФР-1 снижена в результате повышенных уровней ИФР-связывающих белков-1, 2, 4, и 6.
ЗАБОЛЕВАНИЯ ЛЕГКИХ
Любое состояние, сопровождаемое хронической гипоксией, может приводить к задержке роста. Классическими примерами патологии легких, сочетанной с низкорослостью, служат кистозный фиброз и бронхиальная астма, длительно леченная глюкокортикоидами.
ЗАБОЛЕВАНИЯ КРОВИ
Хронические анемии могут сопровождаться задержкой роста ребенка. Нарушенное поступление кислорода в ткани, повышенная нагрузка на сердечнососудистую систему, повышенные энергетические затраты на гемопоэз и нарушение питания рассматривают как основные факторы, вовлеченные в патогенез низкорослости. При B-талассемии описаны нарушенный синтез ИФР-1, гипотиреоз, гипогонадотропный гипогонадизм.
РЕВМАТОИДНЫЙ АРТРИТ
Фармакологические дозы глюкокортикоидов, назначаемые в течение многих лет, связаны с рядом отрицательных побочных эффектов. Эти эффекты включают катаболическое воздействие на белковый обмен, способствуя плохому заживлению ран, повышенной частоте инфекций и повышенной потере костной ткани. Развивается резистентность к инсулину, приводящая со временем к нарушенному углеводному обмену.
При хроническом назначении детям системных глюкокортикоидов побочные эффекты сопровождаются значительным подавлением линейного роста. Задержка роста - частое осложнение ювенильного ревматоидного артрита у детей. Причины задержки роста включают хронический воспалительный процесс, повреждения на уровне кости и суставов, а также нарушение питания. Осложнения активного ювенильного ревматоидного артрита в плане метаболических нарушений и задержки роста усугубляются терапией глюкокортикоидами. Глюкокортикоиды - функциональные антагонисты гормона роста, оказывают влияние как на высвобождение, так и на эффекты гормона роста на органы-мишени. Считается, что глюкокорти-коиды влияют не столько на уровень секреции гормона роста, сколько на профиль ночной секреции, подавляя и задерживая ночные пики секреции гормона роста.
РАХИТ
Недостаток витамина D в организме может быть важной причиной нарушенного роста скелета и в конечном итоге низкорослости. Часто гиповитаминоз D сочетается с другими причинами дефицита роста, такими как неадекватное питание, нарушение всасывания в кишечнике, патология печени и ХПН. При изолированном гиповитаминозе D (следствие пониженного поступления в организм витамина D и отсутствия адекватной инсоляции солнечными лучами) ребенок имеет типичную симптоматику рахита.
Х-сцепленный витамин D-резистентный (гипофосфатемический) рахит развивается вследствие сниженной реабсорбции фосфатов в канальцах почки. Клиническая симптоматика обычно представлена значительным искривлением ног и низкорослостью. Метаболические и скелетные нарушения не купируются приемом витамина D. Лечение включает прием фосфатов внутрь, но такая терапия часто приводит к нарушенной абсорбции кальция в кишечнике. Добавление витамина D к лечению фосфатами повышает абсорбцию кальция в кишечнике и предотвращает гипокальциемию.
ПСЕВДОГИПОПАРАТИРЕОЗ
Задержка роста - частое клиническое проявление манифестации псевдогипопаратиреоза. При классическом варианте заболевания присутствует сочетание низкорослости и характерных дисморфичных черт, гипокальциемии и гиперфосфатемии, вторичной резистентности органов-мишеней к ПТГ. Дети с данным заболеванием, помимо низкорослости, имеют ожирение, округлое лицо, короткие пястные кости, подкожные кальцификаты и задержку умственного развития.
ВРОЖДЕННЫЕ НАРУШЕНИЯ МЕТАБОЛИЗМА
Врожденные нарушения обмена белков, углеводов и липидов (болезнь накопления гликогена, мукополисахаридозы, муколипидозы) часто сопровождаются низкорослостью. Многие врожденные метаболические нарушения ассоциированы со скелетной дисплазией.
ПАТОЛОГИЯ ЭНДОКРИННОЙ СИСТЕМЫ
Гипотиреоз
Постнатальная задержка роста характерна для детей с врожденным гипотиреозом. Неонатальный скрининг на врожденный гипотиреоз способствует ранней диагностике и лечению врожденного гипотиреоза, что предотвращает нарушение процессов роста и развития ребенка.
У ребенка с низким ростом, чей анамнез и антропометрия предполагают дефицит гормона роста, выявление дефицита гормона роста/ИФР-1 требует проведения стимулирующих тестов и определения уровней ИФР-1/ИФР-связывающего белка-3, но после того как исключен гипотиреоз.
Задержка роста может быть симптомом манифестации хронического приобретенного гипотиреоза, причиной которого в большинстве случаев может быть АИТ. Изолированный вторичный и третичный гипотиреоз вследствие дефицита ТТГ и тиролиберина соответственно - крайне редкая причина приобретенного гипотиреоза. ЗГТ левотироксином натрия способствует быстрому скачку роста в начале лечения. Однако данное ускорение роста часто не приводит к полному восстановлению ростового потенциала в большинстве случаев вследствие быстрого прогрессирования скелетного созревания в первые 18 мес лечения.
Болезнь Иценко-Кушинга
Избыток глюкокортикоидов оказывает значительное влияние на процессы роста ребенка, независимо от этиологии гиперкортицизма. Считают, что глюкокортикоиды воздействуют непосредственно на уровне эпифиза, поскольку обычно секреция гормона роста сохранена и уровни ИФР и ИФР-связывающего белка не изменены. Важно отметить, что у детей манифестация болезни Иценко- Кушинга может проявляться исключительно снижением скорости роста, с отсутствием характерных клинических симптомов (ожирение, стрии, снижение мышечной массы, остеопороз).
Сахарный диабет
Задержку роста умеренной степени можно наблюдать у детей с плохо контролируемым СД. Выраженная низкорослость характерна для синдрома Мориака, симптомокомплекса, включающего СД, задержку роста и гепатомегалию вследствие избыточного накопления гликогена в печени. Факторы, вовлеченные в патогенез низкорослости, включают потерю белка, хронический ацидоз, повышенную продукцию глюкокортикоидов, хронический дефицит инсулина. Хронический дефицит инсулина приводит к повышенному уровню ИФР-связывающего белка-1, который, в свою очередь, подавляет эффекты ИФР. Следует отметить, однако, отсутствие жесткой корреляции между контролем гликемии и скелетным ростом. Так, при очевидно неудовлетворительном контроле гликемии многие дети имеют нормальный характер роста. Вероятно, данные пациенты способны поддерживать нормальное внутриклеточное питание тканей, несмотря на явную гиперинсулинемию.
ПСИХОСОЦИАЛЬНЫЙ (ДЕПРИВАЦИОННЫЙ) НАНИЗМ
В основе причин низкорослости могут лежать неблагоприятные условия жизни ребенка, включая социальные условия и неблагополучные семьи (недостаточное питание, постоянные психоэмоциональные стрессы). Для данной категории детей характерно улучшение ростовых процессов при устранении факторов социальной депривации. Нейроэндокринные механизмы депривационного нанизма до конца не изучены. Секреция гормона роста на фоне гормона роста-стимулирующих тестов снижена, секреция АКТГ и ТТГ также может быть снижена. Лечение гормоном роста обычно неэффективно до тех пор, пока не будут исключены неблагоприятные условия жизни ребенка.
СПИСОК ЛИТЕРАТУРЫ
-
Дедов И.И., Тюльпаков А.Н., Петеркова В.А. Соматотропная недостаточность. - М., 1998. - С. 302.
-
Фофанова О.В., Волеводз Н.Н., Безлепкина О.Б. Проект национального консенсуса по диагностике и лечению соматотропной недостаточности у детей. Материалы конференции по детской эндокринологии. - М., 27-28 ноября 2003 г.
-
Consensus guidelines for the diagnosis and treatment of Growth Hormone Deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research Society // J. Clin. Endocrinol. Metab. - 2000. - Vol. 85 (11). - P. 3990-3993.
-
AAcE Medical Guidelines for Clinical Practice: For Growth Hormone Use in Adults and Children - 2003 // Update. Endocr Pract. - 2003. - Vol. 9. - P. 65-76.
-
Saggese G., Ranke M.B., Saenger P. et al. Diagnosis and treatment of growth hormone deficiency in children and adolescents: towards a consensus. Ten years after the availability of recombinant human growth hormone / Workshop held in Pisa, Italy, 27-28 March 1998 // Horm. Res. - 1998. - Vol. 50. - P. 320-340.
-
Fidotti E. A history of growth hormone injections devices // J. Pediatr. Endocrinol. Metab. - 2001. - Vol. 14. - P. 497-501.
АКРОМЕГАЛИЯ И ГИПОФИЗАРНЫЙ ГИГАНТИЗМ
Пронин В. C.
Хроническая гиперпродукция СТГ и ИФР клинически выражается гигантизмом или акромегалией в зависимости от того, когда (до или после завершения пубертата) она развивается.
Гипофизарный гигантизм - клинический синдром, характеризующийся ускоренным линейным ростом с пропорциональным увеличением костей скелета, мягких тканей и возникающий у детей и подростков с незавершенным физиологическим ростом (открытыми зонами роста). Патологическим считается рост, превышающий 97 перцентиль (выше 200 см у мужчин и 190 см у женщин).
Акромегалия - нейроэндокринное заболевание, вызванное хронической избыточной секрецией СТГ у лиц с законченным физиологическим ростом и характеризующееся патологическим диспропорциональным периостальным ростом костей, увеличением размеров мягких тканей и внутренних органов, а также сочетанными системными и обменными нарушениями.
КОД ПО МКБ-10
СКРИНИНГ
Обследуют высокорослых детей и подростков для исключения возможных эндокринных и генетических нарушений, а также взрослых лиц с укрупненными чертами лица, макрогнатией, прогрессирующим увеличением размеров кистей и стоп, сочетанием кардиореспираторных, обменных и множественных неопластических заболеваний.
ЭПИДЕМИОЛОГИЯ
Согласно данным международных регистров, распространенность акромегалии по обращаемости колеблется от 40 до 125 пациентов на 1 млн жителей, тогда как результаты селективного скрининга увеличивают этот показатель до 480- 1034 больных/млн, свидетельствуя о более широком реальном присутствии данной патологии в популяции. Ежегодно регистрируется 3-4 новых случая на 1 млн жителей. Средний возраст пациентов при диагностике заболевания составляет 40-50 лет. Несмотря на небольшую распространенность заболевания, смертность при акромегалии (при отсутствии своевременной диагностики и адекватного лечения) в 2-4 раза превышает таковую в общей популяции, что прямо коррелирует с продолжительностью повышения уровней СТГ и ИФР-I в крови. Среди причин смерти выделяют кардиоваскулярные нарушения (60%), респираторные осложнения (25%), онкологические заболевания (15%). Достижение и поддержание у больных нормальных концентраций СТГ и ИФР-I, коррекция имеющихся органных и обменных нарушений способствуют увеличению выживаемости.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Более чем в 95% случаев у больных акромегалией выявляется доброкачественная моноклональная аденома гипофиза, не окруженная гиперпластической тканью. Как правило, гормон роста-секретирующие аденомы отличаются медленным развитием и длительным скрытым периодом. В 25-45% случаев определяются смешанные аденомы гипофиза, продуцирующие, помимо гормона роста, пролак-тин, ТТГ, АКТГ, ЛГ, ФСГ, α-субъединицу. В редких случаях (менее 2%) причиной акромегалии является эктопическая опухоль, состоящая из нейроэндокринных клеток системы и имеющая эндокраниальное (опухоль глоточного и сфеноидального синуса) или экстракраниальное (опухолевые процессы в легких, средостении, поджелудочной железе, гонадах, кишечнике) расположение. При этом опухолевые клетки продуцируют либо СТГ, либо соматолиберин. Описаны случаи ятрогенной акромегалии, вызванной многолетним злоупотреблением препаратами гормона роста. Примерно 1% СТГ-продуцирующих опухолей приходится на семейные формы и наследственно обусловленные эндокринные нарушения: синдром (МЭН-1), синдром Мак-Кьюн-Олбрайта, комплекс Карни, изолированную семейную акромегалию (табл. 14.10).
| Синдромы генетических нарушений | Клинические проявления |
|---|---|
Синдром множественной эндокринной неоплазии (МЭН-1, синдром Вермера) |
ПГПТ (97% пациентов), островково-клеточные опухоли (80%) и опухоли аденогипофиза (54%). Описаны карциноидные опухоли пищеварительного тракта, множественные лицевые ангиофибромы, коллагеномы, подкожный и висцеральный липоматоз, злокачественные меланомы, лейомиомы пищевода, легких, прямой кишки, матки |
Синдром Мак-Кьюн-Олбрайта |
Преждевременное половое развитие, костно-фиброзная дисплазия, локальная дермопатия (появление на коже пятен цвета кофе с молоком), опухоль гипофиза |
Комплекс Карни |
Смешанные (СТГ-пролактин-секретирующие) опухоли, миксома сердца, пятнистая кожная пигментация, узелковая дисплазия надпочечников, шванномы и т.д. |
Изолированная семейная акромегалия |
Регистрируется при выявлении в семье 2-х или более случаев акромегалии (гигантизма) при отсутствии признаков МЭН-1 или комплекса Карни. Отличается молодым возрастом дебюта заболевания, доминирующим проявлением у мужчин и быстрым ростом аденомы |
Согласно классификации ВОЗ, выделяются различные патоморфологические варианты гормона роста-секретирующих опухолей, отличающиеся по встречаемости и клиническим проявлениям (табл. 14.11).
| Варианты морфологического строения | % встречаемости | Клинические проявления |
|---|---|---|
Соматотропиномы, состоящие из слабо гранулированных хромофобных клеток |
20-30% |
Возникают в молодом возрасте, отличаются высокой гормональной и пролиферативной активностью, склонностью к инвазии и резистентностью к проводимому лечению |
Соматотропиномы, состоящие из густо гранулированных эозинофильных клеток |
20-30% |
Отличаются доброкачественным течением. Манифестируют у лиц старше 50 лет |
Смешанные опухоли (соматопролактиномы, маммосоматотропиномы) |
20-25% |
Отличаются ускоренным и инвазивным ростом с развитием признаков интракраниальной компрессии |
Опухоли из ацидофильных стволовых клеток |
2-5% |
Агрессивное течение, инвазивный рост. Возникают в юношеском возрасте. Проявляются гигантизмом |
Плюригормональные аденомы I и II типа |
2-5% |
Характеризуются инфильтративным ростом и рецидивирующим течением |
Атипичные аденомы, гипофизарные карциномы |
1,5-3% |
Отличаются высокой инвазивностью и быстрым ростом |
КЛИНИЧЕСКАЯ КАРТИНА
Акромегалия является тяжелым полиорганным заболеванием, негативно влияющим на качество и продолжительность жизни пациентов. Этому способствует формирование при акромегалии патологического комплекса прогрессирующих гормональных, метаболических и системных нарушений, каждое из которых является независимым фактором риска ранней инвалидизации и преждевременной смерти.
Клиническая симптоматика акромегалии характеризуется, с одной стороны, признаками компрессионного воздействия опухолевой массы на окружающие ткани (головная боль, сужение полей зрения, гипопитуитаризм, гидроцефалия, гипоталамические расстройства и т.д.), тогда как, с другой - множественными проявлениями системных и обменных нарушений, вызванных продолжительной гиперпродукцией СТГ и ИФР-I.
Больные акромегалией предъявляют жалобы на изменение внешности, увеличение размеров кистей, стоп, головную боль, избыточную потливость, мышечную слабость, онемение пальцев рук и ног, нарушение прикуса, боли в суставах, ухудшение зрения.
При осмотре выявляется характерное укрупнение черт лица за счет увеличения надбровных дуг, скуловых костей, нижней челюсти (прогнатизм), гипертрофии мягких тканей (носа, губ, ушей). Отмечается прогрессирующий рост окружности головы, утолщение пальцев («сигарообразные» пальцы), увеличение размеров кистей и стоп («ластообразные» кисти и стопы). Утолщение голосовых связок и расширение воздушных пазух приводит к появлению характерного низкого грубого голоса.
Кожа плотная, утолщенная, с глубокими складками, более выраженными на волосистой части головы. В области складок и в местах повышенного трения отмечаются зоны гиперпигментации. Нередко выявляют гипертрихоз, гирсутизм, жирную себорею, acne vulgaris, acanthosis nigricans, повышенную влажность кожных покровов (за счет избыточного образования сальных и потовых желез).
Патогномоничным является увеличение клеточной массы всех внутренних органов (спланхномегалия). По мере развития заболевания происходит поступательное замещение функционально-активной ткани на соединительную с развитием полиорганных склеротических изменений (кардиосклероз, пневмосклероз, цирроз печени, фиброз поджелудочной железы и др.). Первично возникающая гипертрофия мышечной ткани в последующем сменяется развитием проксимальной миопатии. Больных беспокоят слабость, снижение толерантности к физическим нагрузкам. Нередко выявляют деформацию позвоночника по типу патологического кифосколиоза с образованием остеофитов и появлением радикулярных болей. Акромегалическая остеоартропатия проявляется дегенеративными изменениями, нарушением подвижности и дестабилизацией суставов.
Легочно-сердечная недостаточность развивается в результате нарушения кровоснабжения и склерозирования гипертрофированных внутренних органов. Характерно наличие эмфиземы легких и пневмосклероза. Вследствие разрастания челюстей и мягких тканей языка и надгортанника у больных нередко возникают обструктивные ночные апное, что в сочетании с прогрессирующей деформацией позвоночника и поражением дыхательной мускулатуры приводит к развитию хронической гипоксии и рестриктивных легочных заболеваний.
Возникновению акромегалической кардиомиопатии способствуют метаболические нарушения, микроангиопатия, гипоксия, а также АГ. У больных формируется концентрическая гипертрофия миокарда, которая в последующем сменяется дилатационной миокардиодистрофией с развитием прогрессирующей сердечной недостаточности. Гипертрофия желудочков приводит к нарушению ритма и проводимости, а также клапанным нарушениям, которые сохраняются, несмотря на ремиссию заболевания. Поражение миокарда усугубляет присоединение атеросклероза коронарных артерий с развитием хронической ишемии. Развитие АГ у больных акромегалией обусловлено задержкой натрия и воды в организме, снижением продукции предсердного натрийуретического пептида, повышением сосудистого тонуса, а также наличием инсулинорезистентности. Часто возникают:
-
диффузный (смешанный) эутиреоидный зоб (выявляется у 76% больных). Для заболевания характерно наличие многоузлового коллоидного в разной степени пролиферирующего зоба. Его патогенез обусловлен избытком СТГ и повышением почечного клиренса для йода;
-
НТГ или манифестный СД, что связано с развитием инсулинорезистентности и гиперинсулинемии в результате прямого липолитического действия гормона роста;
-
патология фосфорно-кальциевого обмена, проявляющаяся гиперкальциурией (часто нефролитиаз) и повышением уровня неорганического фосфора в крови. Потеря кальция с мочой компенсируется ускорением его всасывания через ЖКТ благодаря повышению продукции ПТГ. Также следует помнить о сочетании акромегалии с ПГПТ при синдроме МЭН-1.
Масс-эффект. Интраселлярная компрессия, возникающая по мере увеличения объема опухолевой массы, приводит к формированию парциальной гипофизарной недостаточности с развитием вторичного гипогонадизма, гипотиреоза, гипокортицизма, а также несахарного диабета. Признаки гипопитуитаризма наблюдаются у 40% больных акромегалией.
Выход опухоли за пределы турецкого седла сопровождается широким спектром неврологических, сосудистых и функциональных отклонений, характер которых определяется направлением опухолевого роста.
Супраселлярный рост со сдавлением зрительных нервов (или их перекреста) проявляется развитием хиазмального синдрома, характеризующегося моно-или битемпоральной гемианопсией, прогрессирующим сужением полей зрения, отеком и атрофией дисков зрительных нервов. При окклюзии отверстий Монро развивается окклюзионно-гипертензионный синдром:
-
параселлярное распространение аденомы с прорастанием в кавернозные синусы проявляется поражением III-VI пар черепно-мозговых нервов с развитием птоза, диплопии, дисфункции зрачков, офтальмоплегии, лицевой анальгезии, болезненности по ходу тройничного нерва, снижения слуха;
-
инфраселлярное направление приводит к разрушению дна турецкого седла и прорастанию опухоли в пазуху клиновидной кости, решетчатые пазухи, что может сопровождаться чувством заложенности носа и ликвореей;
-
прорастание аденомы в третий желудочек может вызывать окклюзионную гидроцефалию и несахарный диабет;
-
опухолевая компрессия гипоталамуса и вышележащих отделов ЦНС с нарушением гемо- и ликвородинамики сопровождается появлением аносмии, диэнцефальной эпилепсии, лихорадки, изменений пищевого поведения, формулы сна и бодрствования;
-
при поражении височных долей наблюдаются сложные парциальные припадки, при вовлечении лобных долей - характерные изменения интеллекта и личности, а при локализации опухоли в задней ямке - стволовые нарушения;
-
повышение внутричерепного давления и/или компрессия диафрагмы турецкого седла растущей опухолью обуславливает развитие краниалгий, которые иногда носят упорный характер, доводя больного до исступления. Как правило, головная боль локализуется в лобной, височной, позадиглазничной областях, обычно тупая, не сопровождающаяся тошнотой, не зависящая от положения тела и не всегда снимающаяся аналгезирующими средствами.
Поражение периферической нервной системы обусловлено сдавлением нервных стволов гиперплазированными (и отечными) мягкими тканями и костными образованиями, что проявляется потерей тактильной и болевой чувствительности пальцев рук, развитием парестезий (в результате сдавления срединного нерва в карпальном канале, а также сегментарной демиелинизации периферических нервов). Распространенность карпального туннельного синдрома при акромегалии составляет 20-50%. При МРТ выявлено увеличение размера срединного нерва и повышение интенсивности сигнала, что обусловлено его отеком и повреждением миелиновых оболочек. В ряде случаев у больных с активной акромегалией наблюдается синдром беспокойных ног, приводящий к нарушению сна, дневной сонливости и снижению качества жизни.
Сочетанное пролиферативное и антиапоптотическое действие СТГ и ИФР-I при акромегалии проявляется наклонностью к раннему развитию в организме сочетанных доброкачественных или злокачественных пролиферативных процессов. Среди гастроинтестинальных нарушений при акромегалии следует упомянуть колоректальный рак, полипы и долихоколон. Полипы кишечника выявляются у 45% больных, которые в 24% случаев имеют аденоматозный характер, локализуясь в различных отделах ЖКТ. Независимыми маркерами, ассоциированными с развитием полипов у больных акромегалией, являются хроническое повышение уровня ИФР-I, возраст дебюта и длительность активной стадии. Миомы матки, диффузная или очаговая фиброзно-кистозная мастопатия выявляются при акромегалии в 59 и 58% случаев соответственно.
Распространенность рака простаты, молочных желез, кишечника, легких, щитовидной железы при акромегалии существенно выше, чем в популяции. При акромегалии описаны карциномы толстого кишечника, молочных желез, предстательной железы, бронхов, желудка, пищевода, поджелудочной железы, щитовидной железы, надпочечников, почек, яичников, желчного пузыря, матки. Сравнительное превышение числа предраковых и раковых проявлений у больных акромегалией по сравнению с популяционными значениями указывает на необходимость широкого диагностического исследования и динамического наблюдения.
Как показывает практика, характер клинического течения акромегалии зависит от возраста дебюта заболевания. Чем моложе возраст пациента, тем выше функциональная активность опухоли и тем агрессивнее осуществляется ее рост с выходом за пределы турецкого седла, развитием зрительных и неврологических нарушений. Напротив, если дебют заболевания возникает у лиц старше 50 лет, то опухолевый рост происходит гораздо медленнее и проявляется относительно невысокой гормональной активностью. В этом случае ведущим негативным фактором является большая продолжительность латентного периода, которая приводит к формированию полиорганной недостаточности, негативно влияющей на выживаемость пациентов. Эти особенности развития патологического процесса необходимо учитывать при выборе лечебной стратегии.
ДИАГНОСТИКА
Лабораторные признаки активной стадии акромегалии:
Обычно содержание СТГ определяют утром, натощак, путем 3-кратного взятия порции крови с помощью катетера каждые 20 мин с последующим перемешиванием. Однократное определение базального уровня СТГ не представляет диагностической ценности, поскольку многие стрессовые и метаболические факторы, а также особенность ритмической секреции СТГ могут приводить к получению ложных результатов.
Увеличение содержания в крови ИФР-I с учетом половой и возрастной нормы является достоверным интегральным критерием, указывающим на наличие гиперсекреции СТГ и степень активности акромегалии. Чувствительность метода определения ИФР-I при диагностике акромегалии составляет 100%, специфичность - 97%. Повышение уровня ИФР-I в крови наблюдается во время беременности или экзогенного введения препарата СТГ. Ложное снижение регистрируется при плохо контролируемом СД, недостаточном питании, ХПН, заболеваниях печени, приеме эстрогенных препаратов.
Инструментальные исследования
Для выявления аденом гипофиза и определения особенностей их распространения показано проведение МРТ головного мозга и гипофиза. Обязательным является контрастное усиление с введением парамагнитных контрастирующих веществ (гадопентетовая кислота, гадодиамид), которые с разной скоростью накапливаются в здоровой и опухолево-измененной ткани. Рекомендованная напряженность магнитного поля магнитно-резонансного томографа более 1,5 Тл. Толщина среза в 2 мм является оптимальной для визуализации различных структур гипоталамо-гипофизарной области, позволяя обнаружить микроаденомы размером 2-3 мм.
Как правило, соматотропиномы локализуются в латеральных отделах аденогипофиза. Чаще всего аденомы выглядят гипоили гиперинтенсивными образованиями по отношению к ткани гипофиза на Т1- или Т2-взвешенных изображениях соответственно. У половины микроаденом и трети макроаденом описан феномен резкого снижения сигнала на Т2-взвешенных изображениях, что обусловлено морфологическим различием соматотропином. Неоднородность сигнала от образования связана с наличием кистозной дегенерации или геморрагии. Выделяют микроаденомы (с максимальным диаметром ≤10 мм), макроаденомы (диаметром 11-39 мм) и гигантские аденомы (диаметром ≥40 мм).
Для систематизации используется классификация J. Hardy, которая учитывает степень увеличения опухоли гипофиза согласно данным МРТ:
-
0 степень - интраселлярная микроаденома (≤10 мм в диаметре);
-
1 степень - интраселлярная микроаденома с локальным разрушением турецкого седла;
-
2 степень - интраселлярная макроаденома (>10 мм в диаметре), полностью заполняющая полость турецкого седла;
-
3 степень - макроаденома с локальной деструкцией дна турецкого седла и инвазией сфеноидального или кавернозного синусов;
-
4 степень - макроаденома с тотальным разрушением турецкого седла и локальной инвазией.
Для динамического наблюдения более удобным представляется вычисление объема опухолевой ткани по формуле De Chiro & Nelson:
А×В×С×π/6,
где А - сагиттальный, В - фронтальный, С - аксиальный размеры гипофиза (в см). Микроили макроаденома гипофиза регистрируются при объеме опухоли ≤0,52 см3 или >0,52 см3 .
Офтальмологическое исследование включает осмотр глазного дна и проведение периметрии, что позволяет выявить наличие хиазмального синдрома и патологию дисков зрительных нервов.
Дополнительное обследование. Важным является проведение суточного мониторирования АД и ЭКГ, сомнографии, эхокардиографии, УЗИ щитовидной железы, органов брюшной полости, малого таза, а также колоноскопии с целью своевременного выявления и коррекции осложнений.
Диагноз основан на изучении клинической картины заболевания, определении гормональной и пролиферативной активности СТГ-секретирующей аденомы гипофиза. При этом выделяют активную стадию акромегалии и стадию контроля, под которой понимается стойкая нормализация секреции СТГ, обусловленная хирургическим, фармакологическим, лучевым или комбинированным лечебным пособием (табл. 14.12).
| Стадии | Критерии стадий | Рекомендуемое пособие |
|---|---|---|
Активная стадия |
Спорадический уровень СТГ >1 нг/мл Надир СТГ при ОГТТ ≥0,4 нг/мл Повышенное содержание ИФР-I Клиническая активность заболевания |
Более активное лечение или смена терапии Мониторирование и активное лечение осложнений Ежегодный контроль МРТ |
Стадия контроля акромегалии |
Спорадический уровень СТГ <1 нг/мл Надир СТГ при ОГТТ <0,4 нг/мл Уровень ИФР-I соответствует возрастно-половой норме |
Продолжение лечения, обсуждение возможности снижения дозы аналогов соматостатина Периодическое, но менее частое проведение МРТ (каждые 2-3 года) |
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Акромегалию следует дифференцировать от акромегалоидных состояний (пахидермопериостоза, болезни Педжета, синдрома Мари-Бамбергера), при которых наблюдают сходные внешние проявления при нормальной концентрации СТГ и ИФР-I в крови и при отсутствии опухоли гипофиза. Гигантизм дифференцируют от других форм ускоренного роста, наблюдаемых в перинатальном (синдромы Сотоса, Беквита-Видемана) и постнатальном периодах жизни: семейная (конституциональная) высокорослость, преждевременное половое развитие, первичный или вторичный гипогонадизм, синдромы Марфана и Пайла.
ЛЕЧЕНИЕ
Цели
Ликвидация (или блокирование) источника избыточной продукции СТГ, снижение секреции СТГ и ИФР-I до безопасного уровня, максимально возможное устранение клинических симптомов и признаков заболевания, повышение качества жизни пациентов и профилактика рецидивов заболевания. Первоочередной задачей является скорейшее достижение клинико-биохимической ремиссии как предпосылки для нормализации имеющихся системных изменений и профилактики витальных осложнений.
Показания к госпитализации
-
Больные с активной стадией акромегалии для проведения системного обследования и выработки адекватной лечебной стратегии.
-
Пациенты, прошедшие или находящиеся на лечении с целью оценки его эффективности и динамического обследования.
-
Больные с тяжелой формой акромегалии для лечения соматических осложнений.
Методы лечения
Существуют три метода лечения акромегалии (хирургический, медикаментозный и лучевой), приоритетность использования которых зависит от конкретной клинической ситуации. Факторами, определяющими выбор лечебного пособия, являются размер и характер роста аденомы, состояние зрительных функций, уровни СТГ и ИФР-I, возраст больного, степень тяжести осложнений.
Хирургическое лечение
Основным методом лечения акромегалии является оперативное удаление опухоли гипофиза, позволяющее добиться скорой ремиссии заболевания. Средством выбора при удалении интраселлярных микроаденом и неинвазивных макроаденом является селективная аденомэктомия трансназальным транссфеноидальным доступом. При оперативном удалении микроаденомы полная биохимическая ремиссия (с нормализацией уровня ИФР-I в крови) наблюдается в 75-80% случаев, тогда как радикальное удаление неинвазивной макроаденомы достигается лишь у 40-60% пациентов. Сам факт наличия опухоли больших размеров указывает на техническую сложность достижения клинико-биохимической ремиссии. В 43% случаев сохраняется продолженный рост и в 2-3% случаев - рецидивирующее течение. При инвазивных макроаденомах рекомендуется проведение резекции (не менее 75% от всего опухолевого объема) с последующим назначением лекарственной терапии. К прогностическим факторам неэффективного хирургического пособия относятся: значительные размеры опухоли, ее распространенность в кавернозные синусы и содержание гормона роста более 50 нг/мл.
По данным проведенного мета-анализа, послеоперационная биохимическая ремиссия акромегалии наблюдается при микроаденоме в 75% случаев, при макроаденоме: интраселлярной - в 74%; параселлярной (или в сторону сфеноидального синуса) - в 42%; с супраселлярным ростом (без зрительных нарушений) - в 45%; с супраселлярным ростом и нарушением зрительных функций - в 33%; при гигантской аденоме - в 10% случаев.
Медикаментозное лечение
Показаниями для назначения медикаментозной терапии являются:
-
первичная терапия при бесперспективности или наличии противопоказаний к хирургическому лечению;
-
необходимость предоперационной подготовки для улучшения соматического статуса и снижения риска внутри- и послеоперационных осложнений;
-
обеспечение полного (или частичного) контроля на период после проведенной лучевой терапии и до проявления максимального терапевтического эффекта.
В настоящее время активно используются 3 группы ЛС: аналоги соматостатина (октреотид, ланреотид), агонисты дофамина (бромокриптин, каберголин) и ингибиторы рецепторов СТГ (пегвисомантρ ). Все эти соединения имеют свои терапевтические ниши и используются в качестве моно- (или комбинированной) терапии.
Аналоги соматостатина. В отличие от нативного соматостатина, аналоги соматостатина избирательно связываются со 2-м и 5-м подтипами соматостатиновых рецепторов, контролирующими секрецию гормона роста. Причем их аффинитет ко 2-му лиганду в 10 раз выше, чем к 5-му подтипу соматостатиновых рецепторов. Клетки СТГ-секретирующих аденом гипофиза также преимущественно экспрессируют 2-й и 5-й подтипы соматостатиновых рецепторов, аффинность к которым может отличаться в различных морфологических типах опухолей. Аналоги соматостатина обладают выраженным антисекреторным и антипролиферативным действием, способствуя у чувствительных к препарату больных скорому достижению клинико-биохимической ремиссии и уменьшению объема опухолевой ткани.
К настоящему времени в России зарегистрированы следующие препараты из группы аналогов соматостатина:
-
-
препараты короткого действия для п/к введения - Сандостатин♠ (Новартис Фарма, Швейцария), Октреотид♠ (ЗАО «Фарм-Синтез», Россия) ампулы по 50 и 100 мкг, Октреотид ФСинтез♠ (ЗАО «Ф-Синтез», Россия) ампулы по 50, 100, 300 мкг;
-
препараты продленного действия для в/м введения - Сандостатин ЛАР♠ (Новартис Фарма, Швейцария), Октреотид-депо♠ (ЗАО «Фарм-Синтез», Россия), Октреотид-лонг ФС♠ (ЗАО «Ф-Синтез», Россия) в дозировках 10, 20 и 30 мг /28 дней, в/м;
-
Октреотид короткого действия вводится 3 раза в сутки, п/к. В суточной дозе 200-300 мкг октреотид снижает секрецию гормона роста на 50% и более у 85% больных акромегалией. У большинства пациентов препарат заметно уменьшает выраженность таких симптомов, как головная боль, отечность кожи и мягких тканей, гипергидроз, боль в суставах и парестезии.
Пролонгированное действие октреотид-содержащих препаратов [октреотид (Сандостатин ЛАР♠ ), Октреотид-депо♠ , Октреотид-лонг ФС♠ ] достигается путем включения активного вещества (октреотида) в микросферы (лиофилизированные гранулы), состоящие из особого поли-DL-лактид-когликолид-глюкозного полимера, обеспечивающего постепенное высвобождение препарата из внутримышечного депо. В последующем введенные микросферы подвергаются биодеградации путем гидролиза. Октреотид длительного высвобождения назначают в/м в начальной дозе 20 мг/28 дней. При необходимости дозу увеличивают до 30 мг или уменьшают до 10 мг с прежней частотой введения.
Ланреотид (соматулин аутожель♠ ) 120 мг представляет собой перенасыщенный водный раствор ланреотида и молекул воды с образованием геля. К достоинствам препарата относятся: отсутствие фармакологического носителя, большая длительность действия (до 56 дней) за счет медленной диффузии кристаллов из п/к депо, равномерность фармакокинетического профиля с отсутствием первоначальных пиков гиперконцентрации, наличие готовой лекарственной формы, малый объем вводимого вещества и подкожный способ введения.
Использование аналогов соматостатина в качестве первичной или вторичной медикаментозной терапии способствует нормализации содержания ИФР-I у 67-75% больных, а также статистически значимому уменьшению размеров аденомы гипофиза примерно у 75% больных после годичного курса лечения. Проведенные сравнительные исследования отмечают сходную терапевтическую эффективность данных лекарственных форм при большей продолжительности эффективного действия ланреотида (соматулин аутожель♠ ).
К сожалению, примерно у 30-40% больных акромегалией выявляется первичная резистентность опухолевых клеток к аналогам соматостатина, поэтому для прогнозирования эффективности длительной терапии предварительно рекомендуется провести пробу с октреотидом короткого действия (100 мкг×3 раза в день, п/к в течение 3 дней). Снижение уровня ИФР-I на фоне пробы более 60% от исходного уровня указывает на хороший прогноз длительной медикаментозной терапии. Снижение ИФР-I менее 30% от исходного требует использования иных лечебных схем.
Среди перспективных направлений медикаментозной терапии следует упомянуть внедрение в клиническую практику пасиреотида - аналога соматостатина, обладающего более широким воздействием на соматостатиновые рецепторы, а также создание химерных соединений, способных к комплексному воздействию на соматостатиновые и дофаминовые (Д2 ) рецепторы (допастатинρ ).
Пасиреотид (сигнифор ♠ ) связывается с 1, 2, 3 и 5 подтипами соматостатиновых рецепторов, демонстрируя меньший аффинитет с 2-м подтипом по сравнению с октреотидом и ланреотидом, но большую аффинность с 1, 3 и 5 подтипами соматостатиновых рецепторов. Благодаря этому пасиреотид оказывает значимое антисекреторное и антипролиферативное воздействие у октреотид-резистентных больных. Отмечено, что использование пасиреотида ЛАРρ в дозе 40-60 мг/28 дней в течение года способствовало достижению биохимического контроля у 21% больных из группы ранее плохо компенсированных пациентов при приеме максимальных доз октреотида (сандостатина ЛАР♠ ) (30 мг/28 дней). Несмотря на удовлетворительную переносимость, следует отметить, что у 65% больных, получавших пасиреотид ЛАРρ , наблюдалось нарушение углеводного обмена по сравнению с 18,4% пациентов, получавших октреотид ЛАРρ , что снижает перспективы его широкого использования за пределами резистентной группы.
В настоящее время проходят клинические испытания новые аналоги соматостатина: допастатин ρ (лекарственное вещество, одновременно влияющее на 2-й подтип соматостатиновых рецепторов и Д2 -рецепторы), соматоприм ρ (соединение связывается с 2, 4 и 5 подтипами соматостатиновых рецепторов) и октреолин ρ (оральная форма аналогов соматостатина).
Из группы агонистов дофамина наиболее эффективным является каберголин, оказывающий селективное влияние на Д2 -рецепторы аденоматозных клеток, при назначении которого в дозе от 1 до 3,5 мг в неделю у 60-70% больных со смешанными аденомами (соматопролактиномами, маммосоматотропиномами) наблюдается достоверное снижение уровня ИФР-I, а в 30-50% случаев - его полная нормализация. Уменьшение размеров аденомы гипофиза отмечалось в 55% случаев. Кроме того, данный препарат используется в комбинированной фармакотерапии как дополнительное средство в случае частичной резистентности к аналогам соматостатина.
Блокатор рецепторов гормона роста. С 2000 г. линейка ЛС была дополнена новым препаратом - пегвисомантомρ (сомавертомρ ), который является генноинженерным аналогом эндогенного гормона роста с 9 мутациями. Благодаря произведенным аминокислотным заменам пегвисомантρ путем конкурентного ингибирования блокирует биологическое действие нативного СТГ в периферических тканях и органах, способствуя терапевтическому снижению уровня ИФР-I в сыворотке крови и профилактике осложнений. В результате проведенных клинических исследований (ACROSTADY и GPOS) было отмечено, что использование пегвисомантаρ в течение 12 мес обеспечивало нормализацию уровня ИФР-I у 97% больных и регресс многих клинических симптомов. Отмечены положительные результаты совместного использования аналогов соматостатина (или каберголина) и пегвисомантаρ , позволяющие повысить чувствительность к аналогам соматостатина и уменьшить терапевтическую дозу обоих препаратов. Начальная доза препарата составляет 10 мг/сут, п/к, максимальная суточная доза - 30 мг/сут. В настоящее время разработана пролонгированная форма препарата с частотой введения 80 мг в/м 1 раз в неделю.
При агрессивных и рецидивирующих вариантах опухолевого развития (при неэффективности хирургического или фармакологического контроля) используются цитостатики (темозоломид, эверолимус, препараты интерферона).
Лучевая терапия
Согласно международным рекомендациям, в связи с отсроченным эффектом наступления ремиссии и высоким риском развития осложнений лучевую терапию используют только при неэффективности оперативного и/или медикаментозного лечения. При проведении стереотаксической радиохирургии (гамма-нож, кибер-нож) нормализация уровня ИФР-I наблюдается через 3, 5 и 10 лет у 45, 58 и 86% больных соответственно. Нередко резистентным больным назначают повторные курсы облучения, что существенно повышает риск побочных нарушений. Среди осложнений - гипофизарная недостаточность, лучевые некрозы, зрительные нарушения, вторичные опухоли, снижение когнитивных функций, а также риск цереброваскулярной смертности.
Дальнейшее ведение (табл. 14.13)
МРТ гипофиза |
Ежегодно, при биохимической ремиссии каждые 2 года |
Эхокардиография |
Ежегодно |
Сомнография |
Ежегодно |
УЗИ периферических артерий и вен |
Ежегодно, особенно при гигантизме |
Контроль уровней СТГ, ИФР-1 |
Каждые 3-6 мес |
Определение пролактина, тестостерона, секс-связывающего глобулина, PSA (мужчины) |
Ежегодно, при необходимости определения свободного тестостерона |
Определение ЛГ, ФСГ, пролактина, 17ß-эстрадиола (женщины) |
Ежегодно (или при планировании беременности) |
УЗИ щитовидной железы, органов брюшной полости, малого таза |
Ежегодно |
Денситометрия |
Каждые 2 года (при наличии у пациента остеопении или остеопороза) |
Колоноскопия |
Каждые 5 лет, при сохраняющейся активности акромегалии или наличии изменений - ежегодно |
Опрос качества жизни ACROQOL |
Ежегодно |
СПИСОК ЛИТЕРАТУРЫ
-
Rosario P.W. Frequency of acromegaly in adults with diabetes or glucose intolerance and estimated prevalence in the general population // Pituitary. - 2011. - Vol. 14 (3). - P. 217-221.
-
Schneider H.J. High prevalence of biochemical acromegaly in primary care patients with elevated IGF-1 levels // Clin. Endocrinol. (Oxf). - 2008. - Vol. 69 (3). - P. 432-435.
-
Dekkers O.M., Biermasz N.R., Pereira A.M. et al. Mortality in acromegaly: a metaanalysis // J. Clin. Endocrinol. Metab. - 2008. - Vol. 93. - P. 61-67.
-
Holdaway I.M., Bolland M.J., Gamble G.D. A meta-analysis of the effect of lowering serum levels of GH and IGF-I on mortality in acromegaly // Eur. J. Endocrinol. - 2008. - Vol. 159. - P. 89-95.
-
Obary A., Sano T., Ohyama K. et al. Clinicopathological features of growth hormone-producing pituitary adenomas: difference among various types defined by cytokeratin distribution pattern including a transitional form // Endocr. Pathol. - 2008. - Vol. 19, N 2. - P. 82-91.
-
Saeger W., Ludecke D.K., Buchfelder M. et al. Pathogistological classification of pituitary tumors: 10 years of experience with the German Pituitary Tumor Registry // Eur. J Endocrinol. - 2007. - Vol. 156. - P. 203-216.
-
Petersenn S., Buchfelder M., Gerbert B. еt al. Age and sex as predictors of biochemical activity in acromegaly: analysis of 1485 patients from the German Acromegaly Register // Clin. Endocrinol. (Oxf). - 2009. - Vol. 71. - N 3. - P. 400-405.
-
Bhansali A., Upreti V., Dutta P. et al. Adolescent acromegaly: clinical parameters and treatment outcome // J. Pediatr. Endocrinol. Metab. - 2010. - Vol. 23. - N 10. - P. 1047-1054.
-
Melmed S., Colao A., Barkan A. et al. Guidelines for acromegaly management: an update // J. Clin. Endocrinol. Metab. - 2009. - Vol. 94. - P. 1509-1517.
-
Sala E., Ferrante E., Locatelli M. et al. Diagnostic features and outcome of surgical therapy of acromegalic patients: experience of the last three decades // Hormones (Athens). - 2014. - Vol. 13 (1). - P. 95-103.
-
Григорьев А.Ю., Азизян В.Н. Эндоскопическая хирургия аденом гипофиза: Практическое руководство для врачей / под ред. И.И. Дедова. - М., 2011.
-
Katznelson L., Atkinson J.L.D., Cook D.M. et al. American Association of clinical endocrinologists medical guidelines for clinical practice for the diagnosis and treatment of acromegaly // Endocrine Practice. - 2011. - Vol. 17 (4). - P. 1-44.
-
Casarini A.P., Jallad R.S., Pinto E.M. et al. Acromegaly: correlation between expression of somatostatin receptor subtypes and response to octreotidelar treatment // Pituitary. - 2009. - Vol. 12 (4). - P. 297-303.
-
Cuevas-Ramos D., Feseriu M. Somatostatin receptor ligands and resistance to treatment in pituitary adenomas // J.Mol. Endocrinol. - 2014. - Vol. 52 (3). - P. R223-R240.
-
Plöckinger U., Albrecht S., Mawrin C. et al. Selective Loss of Somatostatin Receptor 2 in Octreotide-Resistant Growth Hormone-Secreting Adenomas // J. Clin. Endocrinol. Metab. - 2008. - Vol. 93 (4). - P. 1203-1210.
-
Пронин В.С., Потешкин Ю.Е., Гитель Е.П. Современная стратегия диагностики и лечения соматотропином. - М.: ГЕОТАР-Медиа, 2013. - 191 с.
-
Gadelha M., Bronstein M.D., Brue T. et al. Pasireotide LAR demonstrates superior efficacy versus octreotide LAR or lanreotide ATG in patients with inadequately controlled acromegaly: results from a phase III, multicentre, randomized study (PAOLA) 16th European Congress of Endocrinology (ECE) 2014 3-7 May, Wroslaw, Poland // Endocrine Abstract. - 2014. - Vol. 35. - P. 907.
-
Breitschaft A., Hu K., Hermosilo Resendiz K. et al. Management of hyperglycemia associated with pasireotide (SOM230): healthy volunteer study // Diabetes Res. Clin. Pract. - 2014. - Vol. 103 (3). - P. 458-465.
-
Vilar L., Czepielewsk M.A., Naves L.A. et al. Substantial shrinkage of adenomas cosecreting growth hormone and prolactin with use of cabergoline therapy // Endocr. Pract. - 2007. - Vol. 13 (4). - P. 396-402.
-
Marazuela M., Ramos-Levi A., Sampedro-Nunez M., Bernabeu I. Cabergoline treatment in acromegaly // Endocrine. - 2014.
-
van der Lely A.J., Biller B.M., Brue T. et al. Long-term safety of pegvisomant in patiens with acromegaly: comprehensive review of 1288 subjects in ACROSTUDY // J. Clin. Endocrinol. Metab. - 2012. - Vol. 97. - P. 1589-1597.
-
Losa M., Piani C., Mortini P. Medical treatment of aggressive pituitary tumors: temozolomide 2nd Workshop Aggressive pituitary tumors. - Munich, Germany, December 1-3, 2011. - P. 32.
-
Del Porto L.A., Liubinas S.V., Kaye A.H. Treatment of persistent and recurrent acromegaly // J. Clin. Neurosci. - 2011. - Vol. 18 (2). - P. 181-190.
-
Carrasco C.A., Gadellaq M., Manavela M. et al. Aggressive tumors and difficult choices in acromegaly // Pituitary. - 2014. - Vol. 17 (Suppl. 1). - P. 24-29.
-
Barkan A.L. Radiotherapy in acromegaly: the argument against // Clin. Endocrinol. - 2003. - Vol. 58. - P. 132-135.
-
Melmed S., Bonert V., Freseriu M., Kleinberg D.L. Acromegaly: Assessing the Disorder and Navigating Therapeutic Options for Treatment // Endocrine Practice. - 2014. - Vol. 20 (suppl. 1).
БОЛЕЗНЬ ИЦЕНКО-КУШИНГА
Иловайская И.А.
СИНОНИМЫ
Болезнь Кушинга, АКТГ-зависимый гиперкортицизм, болезнь Иценко-Кушинга гипофизарного происхождения, гиперкортицизм центрального происхождения.
ОПРЕДЕЛЕНИЕ
Болезнь Иценко-Кушинга - тяжелое заболевание, сопровождающееся появлением множества специфических симптомов и развивающееся вследствие повышенной продукции гормонов коры надпочечников, что обусловлено избыточной секрецией АКТГ клетками гиперплазированной или опухолевой ткани гипофиза.
КОД ПО МКБ-10
ЭПИДЕМИОЛОГИЯ
Распространенность АКТГ-зависимого гиперкортицизма составляет 4-5 случаев на 1 млн человек, ежегодно выявляют 1-2 новых случая на 1 млн. Чаще данное заболевание поражает женщин в возрасте 25-40 лет, однако может встречаться и в других возрастных группах. Соотношение заболеваемости женщин и мужчин - от 3:1 до 8:1.
ПРОФИЛАКТИКА
Методы профилактики не разработаны.
СКРИНИНГ
Методы скрининга не разработаны.
КЛАССИФИКАЦИЯ
Выделяют:
ЭТИОЛОГИЯ
Наиболее часто (в 80-85% случаев) причиной гиперкортицизма центрального происхождения служит АКТГ-продуцирующая опухоль гипофиза (кортикотропинома). В подавляющем большинстве случаев такие опухоли носят доброкачественный характер, описано всего 20 случаев АКТГ-секретирующих карцином гипофиза. Соотношение частот, с которыми диагностируют микро- и макроаденомы, составляет 4:1. В некоторых случаях размеры кортикотропиномы могут быть <2 мм, когда опухоль не визуализируется при МРТ, однако ее наличие подтверждают при гистологическом послеоперационном исследовании.
В 10-15% случаев у больных с АКТГ-зависимым гиперкортицизмом опухоль не обнаруживают, а по данным гистологического исследования удаленной ткани гипофиза выявляют гиперплазию кортикотрофов.
ПАТОГЕНЕЗ
Существуют «гипофизарная» и «гипоталамическая» теории развития болезни Иценко-Кушинга. В первом случае предполагают, что причиной заболевания служит мутация кортикотрофов, которая приводит к образованию АКТГ-синтезирующей опухоли гипофиза, функционирующей независимо от гипо-таламических воздействий. Эту теорию подтверждает наличие у большинства больных кортикотропиномы, моноклональное строение опухоли и снижение концентрации кортиколиберина. Во втором случае предполагают, что к гиперсекреции кортиколиберина, обуславливающей гиперстимуляцию кортикотрофов и гиперплазию гипофиза (или формирование вторичной опухоли), приводят гипоталамические нарушения. Не исключено, что оба этих механизма вовлечены в патогенез болезни Иценко-Кушинга.
При болезни Иценко-Кушинга нарушен нормальный циркадный ритм секреции АКТГ. Частота эпизодов (пиков) секреции АКТГ сохранена, однако повышены их амплитуда и длительность. Отсутствует четкая синхронизация секреторной динамической активности АКТГ и кортизола, что, вероятно, подтверждает тот факт, что секреция АКТГ не контролируется гипоталамусом и не подавляется высокими концентрациями периферических глюкокортикоидов по принципу отрицательной обратной связи. Высокие концентрации АКТГ (изолированно или вместе с другими ростовыми факторами) воздействуют на клетки коры надпочечников, вызывая их гиперплазию и гиперсекрецию глюкокортикоидов. Вследствие этого утрачивается и нормальный ритм секреции глюкокортикоидов. Так как активность ферментов, вовлеченных в процесс биосинтеза кортизола, повышается равномерно, увеличиваются концентрация и экскреция не только кортизола, но и всех его предшественников. Секреция АКТГ обычно усиливается в большей степени по сравнению с кортизолом: если продукция АКТГ при болезни Иценко-Кушинга увеличивается в 18-20 раз, то продукция кортизола - всего в 7-10 раз. Возможно, надпочечники частично резистентны к повышенному содержанию АКТГ. Содержание АКТГ и кортизола в плазме крови в утренние часы может быть в пределах физиологической нормы, однако концентрация этих гормонов вечером всегда повышена. Вследствие избыточной продукции кортизола повышается суммарная суточная экскреция свободного кортизола с мочой. Хроническая гиперкортизолемия подавляет секрецию кортиколиберина гипоталамусом и ингибирует продукцию АКТГ нормальными кортикотрофами, что приводит к их атрофии.
При развитии макроаденом чаще, чем при микроаденомах, выявляют повышение концентрации АКТГ (83 и 45% случаев соответственно), при этом высокие концентрации более устойчивы к подавлению большими дозами дексаметазона.
Эффекты больших доз кортизола на организм описаны в разделе «Синдром гиперкортицизма».
КЛИНИЧЕСКАЯ КАРТИНА
ДИАГНОСТИКА
Установить диагноз гиперкортицизма достаточно трудно, так как многие симптомы (например, увеличение массы тела, повышение АД, общая слабость) неспецифичны и встречаются при многих заболеваниях. Тем не менее тщательный сбор анамнеза и физикальное обследование помогают заподозрить гиперкортицизм и предположить его этиологию.
Анамнез и физикальное обследование
См. «Синдром гиперкортицизма».
Лабораторные исследования
См. «Синдром гиперкортицизма».
Инструментальные исследования
Для топической диагностики используют:
Для уточнения состояния органов и систем применяют:
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Описание методов дифференциальной диагностики АКТГ-зависимых и АКТГ-независимых форм гиперкортицизма дано в разделе «Синдром гиперкортицизма».
Если подтверждено наличие АКТГ-зависимого гиперкортицизма, необходимо уточнить источник гиперсекреции АКТГ (АКТГ-секретирующая опухоль гипофиза или эктопическая продукция АКТГ внегипофизарной опухолью).
ПОКАЗАНИЯ К КОНСУЛЬТАЦИИ ДРУГИХ СПЕЦИАЛИСТОВ
С целью оптимального выбора тактики лечения необходимы консультации:
ПРИМЕР ФОРМУЛИРОВКИ ДИАГНОЗА
Болезнь Иценко-Кушинга средней степени тяжести. Микроаденома гипофиза (кортикотропинома). Симптоматическая АГ. Вторичный остеопороз. Аменорея II.
ЛЕЧЕНИЕ
Цели лечения
Показания к госпитализации
Хирургическое лечение
Методом выбора лечения АКТГ-продуцирующих опухолей гипофиза считают селективную аденомэктомию, выполненную высококвалифицированным нейрохирургом.
После нейрохирургического лечения ремиссию заболевания наблюдают в 66-89% случаев, наилучшие результаты наблюдают в специализированных центрах, где специалисты имеют достаточный опыт выполнения подобных операций у данного контингента больных. Оперативное лечение обычно проводят при помощи современных эндоскопических и (или) нейронавигационных нейрохирургических методик трансназальным (транссфеноидальным) доступом.
Основными предикторами успешного исхода оперативного лечения выступают:
Основными критериями успешно выполненного оперативного лечения служат:
Для компенсации НН, возникшей после оперативного лечения, используют препараты гидрокортизона. Принципы лечения такие же, как при вторичной НН. Дозу подбирают индивидуально на основании клинических симптомов. Необходимость приема глюкокортикоидов сохраняется в течение нескольких месяцев после операции.
Снижение концентрации АКТГ и улучшение самочувствия пациента без проявлений НН в раннем послеоперационном периоде в большинстве случев свидетельствуют о недостаточной радикальности проведенного оперативного лечения и о необходимости дальнейшего лечения пациента.
Даже после радикально выполненной операции рецидив заболевания в течение последующих нескольких лет обычно наблюдают примерно у 10% больных.
Лучевое лечение
В качестве второй линии терапии (иногда - как первую линию лечения) у пациентов с болезнью Иценко-Кушинга можно проводить облучение гипофизарной области. Данный вид лечения используют в первую очередь у пациентов:
Наиболее успешно радиохирургическое лечение. В отечественной практике при лечении болезни Иценко-Кушинга облучением пучком протонов ремиссию заболевания отмечают у 88-92% пациентов в возрасте до 20 лет и у 75-85% пациентов среднего возраста. У пациентов в возрасте 40-45 лет эффективность облучения несколько ниже.
После облучения улучшение гормонального статуса и состояния больного отмечают уже через 2-3 мес, ремиссия заболевания наступает через 6-12 мес. В период после облучения и до формирования стойкой ремиссии заболевания возможно применение ингибиторов стероидогенеза для оптимального контроля над продукцией кортизола.
Медикаментозное лечение
Возможно проведение патогенетической и симптоматической медикаментозной терапии при болезни Иценко-Кушинга.
Патогенетическая терапия. В исследованиях in vitro АКТГ-продуцирующих опухолей гипофиза были обнаружены соматостатиновые рецепторы 1 и 5 подтипов, что дает возможность применять аналоги соматостатина последнего поколения в лечении болезни Иценко-Кушинга. Пасиреотид является новым аналогом соматостатина, связывающимся с 1, 2, 3 и 5 подтипом соматостатиновых рецепторов, и в России в конце 2013 г. он был зарегистрирован для терапии врослых пациентов с болезнью Иценко-Кушинга, у которых нейрохирургическое лечение было неэффективно или невыполнимо. По данным крупного многоцентрового рандомизированного исследования III фазы было показано, что на фоне лечения пасиреотидом в дозах 600 или 900 мкг 2 раза в день подкожно примерно у 50% пациентов отмечается значительное снижение уровня свободного кортизола в моче, уровня АКТГ, а также уменьшается выраженность основных клинических проявлений гиперкортицизма. Нормализация уровня свободного кортизола отмечалась у 20% больных, в первую очередь с исходным превышением уровня свободного кортизола не более 5 раз от верхней границы референсных значений. Кроме того, у ряда больных отмечалось уменьшение размеров кортикотропиномы. Побочные эффекты пасиреотида аналогичны побочным эффектам других аналогов соматостатина (октреотида и ланреотида), кроме гипергликемии, которая отмечается у 78% пациентов. На сегодняшний день этот препарат является единственным зарегистрированным ЛС для лечения пациентов с болезнью Иценко-Кушинга.
Кроме того, на клетках кортикотропины обнаруживаются дофаминовые рецепторы, поэтому теоретически возможно применение агонистов дофамина (каберголина). Препарат для лечения болезни Иценко-Кушинга не зарегистрирован, однако в исследованиях было показано, что на фоне применения каберголина 1-7 мг/нед в течение 12 мес отмечается нормализация уровня св. кортизола в моче примерно у 40% пациентов. В настоящее время идет исследование II фазы с целью оценки эффективности и безопасности каберголина при болезни Иценко-Кушинга.
В качестве симптоматической терапии применяют ингибиторы стероидогенеза (табл. 14.14), за счет снижения синтеза кортизола происходит снижение клинических проявлений гиперкортицизма и улучшение общего состояния пациента. Дозу препарата подбирают таким образом, чтобы нормализовать суточную экскрецию свободного кортизола с мочой. Эффект от проводимого лечения временный, отмечается только на фоне приема препаратов. Такое лечение можно рекомендовать пациентам с болезнью Иценко-Кушинга, готовящимся к хирургическому или лучевому лечению. В случае проведения лучевого лечения снижение содержания АКТГ происходит не сразу (в течение нескольких месяцев), поэтому ингибиторы стерои-догенеза назначают до облучения и рекомендуют продолжать их прием в течение 3-6 мес после облучения. Отменяют терапию в том случае, если содержание свободного кортизола в суточной моче становится меньше нижнего уровня референтных значений или если появляются клинические признаки НН.
| Группа ЛС | Название препарата | Путь введения | Разовая доза, мг | Кратность, раз в сутки | Длительность приема |
|---|---|---|---|---|---|
Противоопухолевое средство |
Аминоглутетимид |
Внутрь |
250-500 |
3-4 |
До ликвидации источника гиперпродукции АКТГ |
Ингибитор стероидогенеза в надпочечниках |
Кетоконазол |
Внутрь |
400 |
1-4 |
Кроме ингибиторов стероидогенеза, для симптоматического лечения болезни Иценко-Кушинга применяют антагонист глюкокортикоидных рецепторов мифепристон, который блокирует эффекты кортизола путем связывания с глюкокортикоидными рецепторами 2-го типа (аффинность в 3-4 раза выше, чем у дексаметазона, и в 18 раз выше, чем у кортизола). Терапия мифепристоном сопровождается улучшением клинического состояния пациентов с болезнью Иценко-Кушинга, улучшение углеводного обмена отмечается у 60% и снижение АД у 38% пациентов, снижается масса тела. Однако на терапии мифепристоном наблюдается увеличение уровней АКТГ и свободного кортизола в моче. Связанные с лечением нежелательные явления включают гипокалиемию (44%) и утолщение эндометрия (20%).
Также в симптоматическом лечении болезни Иценко-Кушинга используют препараты различных фармакологических групп в зависимости от клинической симптоматики.
Показания к консультации других специалистов
В зависимости от клинических проявлений заболевания могут потребоваться консультации кардиолога, уролога, гинеколога или андролога, невролога.
Примерные сроки нетрудоспособности
В подавляющем большинстве случаев пациенты с длительным анамнезом и (или) быстро прогрессирующим течением заболевания становятся нетрудоспособными. Восстановление трудоспособности происходит в течение 3-12 мес после патогенетического лечения.
Дальнейшее ведение
За пациентом наблюдают:
ПРОГНОЗ
Без лечения половина пациентов с выраженными клиническими проявлениями гиперкортицизма погибает в течение 5 лет от начала заболевания. Своевременная диагностика и адекватное лечение существенно улучшают прогноз заболевания и качество жизни пациентов.
СПИСОК ЛИТЕРАТУРЫ
-
Болезнь Иценко-Кушинга / Под ред. И.И. Дедова, Г.А. Мельниченко. - М.: УП Принт, 2012. - 342 с.
-
Григорьев А.Ю., Иващенко О.В., Мельниченко Г.А., Марова Е.И., Воронцов А.В., Владимирова В.П., Колесникова Г.С. Хирургическое лечение пациентов с болезнью Иценко-Кушинга // Эндокринная хирургия. - 2007. - №1. - С. 24-27.
-
Colao A., Petersenn S., Newell-Price J., Findling J.W., Gu F., Maldonado M., Schoenherr U., Mills D., Salgado L.R., Biller B.M.;Pasireotide B2305 Study Group. A 12-month phase 3 study of pasireotide in Cushing?s disease // N. Engl. J. Med. - 2012 Mar 8. - Vol. 366 (10). - P. 914-924.
-
Hamrahian A. H., Yuen K. C., Hoffman A. R., For The Aace Neuroendocrine And Pituitary Scientific Committee. AACE/ACE Disease State Clinical Review: Medical Management of Cushing Disease // Endocr. Pract. - 2014 Jul 1. - Vol. 20 (7). - P. 746-757.
-
Hofland L.J. Somatostatin and somatostatin receptors in Cushing?s disease // Mol. Cell Endocrinol. - 2008 May 14. - Vol. 286 (1-2). - P. 199-205.
-
Mazzuco T.L., Bourdeau I., Lacroix A. Adrenal incidentalomas and subclinical Cushing?s syndrome: diagnosis and treatment // Curr. Opin. Endocrinol. Diabetes Obes. - 2009 Jun. - Vol. 16 (3). - P. 203-210.
-
McKeage K. Pasireotide: a review of its use in Cushing?s disease // Drugs. - 2013 May. - Vol. 73 (6). - P. 563-574.
-
Nieman L.K. Update in the medical therapy of Cushing?s disease // Curr. Opin. Endocrinol. Diabetes Obes. - 2013 Aug. - Vol. 20 (4). - P. 330-334.
-
Shimon I., Rot L., Inbar E. Pituitary-directed medical therapy with pasireotide for a corticotroph macroadenoma: pituitary volume reduction and literature review // Pituitary. - 2012 Dec. - Vol. 15 (4). - P. 608-613.
-
Tritos N.A., Biller B.M. Cushing?s disease // Handb. Clin. Neurol. - 2014. - Vol. 124. - P. 221-234.
-
Torales J., Halperin I., Hanzu F., Mora M., Alobid I., De Notaris M., Ferrer E., Enseñat J. Endoscopic endonasal surgery for pituitary tumors. Results in a series of 121 patients operated at the same center and by the same neurosurgeon // Endocrinol. Nutr. - 2014 Oct. - Vol. 61 (8). - P. 410-416.
-
Wilson P.J., Williams J.R., Smee R.I. Cushing?s disease: a single centre?s experience using the linear accelerator (LINAC) for stereotactic radiosurgery and fractionated stereotactic radiotherapy // J. Clin. Neurosci. - 2014 Jan. - Vol. 21 (1). - P. 100-106.
ГОРМОНАЛЬНО-НЕАКТИВНЫЕ ОПУХОЛИ ГИПОФИЗА
Иловайская И.А.
СИНОНИМЫ
«Клинически неактивные» опухоли гипофиза, «неактивные» опухоли гипофиза, «нефункционирующие» опухоли гипофиза.
ОПРЕДЕЛЕНИЕ
ГНОГ - собирательное понятие, включающее различные по морфофункциональным характеристикам типы опухолей гипофиза, объединенные отсутствием явных клинических проявлений эндокринных расстройств, указывающих на избыточную гормональную секрецию. У большинства таких опухолей сохраняется способность секретировать гормоны, выявляемые in vitro.
ЭПИДЕМИОЛОГИЯ
В общей популяции частота ГНОГ составляет примерно 50 случаев на 1 млн населения. Эти опухоли чаще выявляют у пациентов среднего возраста (между 40 и 50 годами жизни), и с одинаковой частотой отмечают у мужчин и женщин. На долю ГНОГ приходится 25-43% всех гипофизарных опухолей.
ПРОФИЛАКТИКА
Профилактика отсутствует.
СКРИНИНГ
Скрининг проводят:
КЛАССИФИКАЦИЯ ОПУХОЛЕЙ ГИПОФИЗА
Классификация по способности продуцировать гормоны in vivo и in vitro:
По спектру гормональной активности выделяют:
Классификация по размеру опухоли (для опухолей гипофиза с любой гормональной активностью):
Классификация по характеру распространения (для опухолей гипофиза с любой гормональной активностью):
Классификация по морфофункциональным особенностям (для ГНОГ):
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Возникновение опухолей гипофиза носит в большинстве случаев спорадический характер, но может быть и наследственно обусловленным. В результате мутаций (активации клеточных протоонкогенов или утраты генов туморсупрессоров, табл. 14.15) образуется аномальная гипофизарная клетка, дающая моноклональный рост опухоли.
| Ген | Тип мутации | Тип опухоли |
|---|---|---|
Активация протоонкогенов |
||
Gsα |
Герминативные и соматические активирующие мутации |
Соматотрофные опухоли |
RAS |
Соматические активирующие мутации |
Карциномы гипофиза |
PKC |
Соматические активирующие мутации |
Соматотрофные опухоли |
PRKAR1 |
Мутации с преждевременной остановкой трансляции |
Соматотрофная аденома (комплекс Карней) |
Ptd-FGFR4 |
Альтернативная транскрипционная инициация |
Все типы опухолей гипофиза |
PTTG1 |
Неизвестен |
Все типы опухолей гипофиза |
Тумор-супрессорные гены |
||
MEN1 |
Инактивирующие мутации |
Все типы опухолей гипофиза |
RB |
Промотер метилации |
Все типы опухолей гипофиза |
CDKN2A |
Промотер метилации |
Все типы опухолей гипофиза |
CDKN1B |
Сниженная экспрессия, неизвестный механизм |
Кортикотрофные и рецидивирующие опухоли |
Изменения рецепторов и ответной реакции трансформированной клетки приводят к изменению ее чувствительности к гипоталамическому контролю, поэтому секреторная и пролиферативная активность аденоматозных клеток в большинстве случаев не зависит от физиологического регулирования и приобретает патологический характер. Нарушается механизм обратной отрицательной связи: различные гипоталамические либерины и статины, факторы роста и гормональные субстанции, не оказывающие влияния на рост нормальных гипофизарных клеток, могут стимулировать развитие аденоматозного клона. В клетках гипофизарных опухолей обычно наблюдают дисрегуляцию протеинов, контролирующих клеточный цикл, в частности выключение генов-ингибиторов пролиферации.
Иммуногистохимические исследования опухолевой ткани in vitro показывают, что 45-85% ГНОГ сохраняют способность продуцировать гормоны: может наблюдаться секреция гонадотропинов в сочетании с общей α-субъединицей гликопротеиновых гормонов (α-СЕ); реже отмечают секрецию пролактина, СТГ, АКТГ.
Возможные причины отсутствия клинических проявлений гормональной активности ГНОГ:
Зависимости клинических признаков от гистопатологических особенностей опухолей в большинстве случаев не выявляют.
ГНОГ не сопровождаются симптомами, характерными для избыточной гормональной секреции, однако вследствие сдавления опухолью нормальной гипофизарной ткани или ножки гипофиза могут сопровождаться проявлениями гипофизарной недостаточности различной степени тяжести и/или умеренной гиперпролактинемией. При распространении опухоли в кавернозные синусы может произойти сдавление III, IV, VI пары черепных нервов, а также 1-й и 2-й ветвей тройничного нерва, что проявляется соответствующими невралгиями, а также провоцирует головные боли. При размерах опухоли более 30 мм может отмечаться сдавление дна III желудочка головного мозга с формированием внутренней гидроцефалии.
КЛИНИЧЕСКАЯ КАРТИНА
Основные жалобы у больных с ГНОГ:
Часто неспецифические жалобы маскируют проявления гипофизарной недостаточности, которые имеют до 60% больных ГНОГ.
В редких случаях первыми клиническими проявлениями заболевания становятся кровоизлияние в опухоль или апоплексия гипофиза (острый геморрагический инфаркт). Для этого состояния характерны внезапная сильная головная боль, тошнота, рвота, может быть офтальмоплегия, нарушения зрения, симптомы острой гипофизарной недостаточности и нарушение сознания различной степени выраженности. В тех случаях, когда кровь проникает в ликвор, можно наблюдать менингиальные стигмы. При этом на МРТ визуализируется резко интенсивный очаг, обусловленный присутствием метгемоглобина.
ДИАГНОСТИКА
Анамнез
При сборе анамнеза следует обратить внимание на возможные первые признаки
ГНОГ:
В некоторых редких случаях первыми симптомами могут быть жажда и/или ликворея.
Физикальное обследование
При физикальном обследовании можно обнаружить симптомы гипопитуитаризма (см. «Синдром приобретенного гипопитуитаризма у взрослых»).
Лабораторные исследования
Для уточнения/исключения гормональной гиперсекреции:
При подозрении на гипопитуитаризм проводят тесты для его исключения: см. «Синдром приобретенного гипопитуитаризма».
Инструментальные обследования
Для уточнения состояния хиазмально-селлярной области:
В отдельных случаях можно рассмотреть вопрос о проведении ПЭТ (с 123 I-epidepride, 123 I-Tyr-octreotide) для выбора дальнейшего медикаментозного лечения.
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Дифференциальную диагностику проводят с:
Часто решающим в проведении дифференциальной диагностики оказывается заключение послеоперационного гистологического исследования удаленной ткани.
ПОКАЗАНИЯ К КОНСУЛЬТАЦИИ ДРУГИХ СПЕЦИАЛИСТОВ
Рекомендуют консультации следующих специалистов:
-
офтальмолога: исследование глазного дна, полей зрения, особенно при супраселлярном распространении опухоли;
-
нейрохирурга (особенно при экстраселлярном распространении опухоли и/ или увеличении размеров опухоли при динамическом наблюдении, в случае клинических признаков апоплексии гипофиза);
-
невролога (особенно при распространении опухоли в кавернозные синусы).
ПРИМЕР ФОРМУЛИРОВКИ ДИАГНОЗА
Эндоселлярная микроаденома гипофиза без признаков гормональной активности.
Эндопараселлярная опухоль гипофиза (гормонально-неактивная). Частичный гипопитуитаризм (вторичный гипогонадизм, вторичный гипотиреоз).
ЛЕЧЕНИЕ
Цели лечения
Цели лечения:
Показания к госпитализации
Госпитализация показана при:
Немедикаментозное лечение
Нередко наиболее приемлемая тактика для больных с ГНОГ - динамическое наблюдение. В большинстве случаев ГНОГ растут крайне медленно и клиническая симптоматика не проявляется в течение длительного периода времени. Обычно отрицательную динамику выявляют в 5% случаев среди микроаденом и в 25% случаев - среди макроаденом.
-
Пациентам, не подлежащим нейрохирургическому лечению, выполняют МРТ головного мозга через 6 мес от выявления при макроаденоме и через 12 мес при микроаденоме (уровень доказательности 1); если опухоль не изменилась в размерах - МРТ повторяют ежегодно при макроаденомах и через 1-2 года при микроаденомах, при отсутствии динамики роста через 3 года период между исследованиями можно увеличить (уровень доказательности 2).
-
Исследование полей зрения проводят у всех пациентов с опухолью гипофиза, близко расположенной к хиазме по данным МРТ (уровень доказательности 1), не проводят пациентам без прилежания опухоли гипофиза к хиазме и/ или без новых клинических симптомов (уровень доказательности 2).
-
Клинические и биохимические исследования для исключения гипопитуитаризма проводят у пациентов с макроаденомой через 6 мес после диагностики и затем ежегодно, хотя обычно симптомы гипопитуитаризма развиваются при увеличении опухоли в размерах (уровень доказательности 1). Обследования для исключения гипопитуитаризма можно не проводить пациентам с микроаденомами гипофиза, без новых клинических симптомов и без отрицательной динамики размеров опухоли по данным МРТ (уровень доказательности 2В).
-
Если у пациентов развиваются какие-либо симптомы, потенциально связанные с опухолью гипофиза, или отмечается увеличение размеров опухоли по данным МРТ, необходимо провести соответствующее клиническое обследование (уровень доказательности 1).
Хирургическое лечение
Хирургическое лечение - основной метод лечения ГНОГ, особенно у пациентов молодого возраста, с высоким риском прогрессирования опухоли или при отрицательной динамике - увеличении размеров опухоли за время наблюдения.
Пациентам с ГНОГ проводят нейрохирургическое лечение в следующих случаях (уровень доказательности 1):
Эффективность оперативного лечения зависит от размеров и характера распространенности опухоли: чем больше размеры опухоли и ее инвазия в окружающие структуры, тем менее вероятно радикальное удаление. Радикальное удаление опухоли удается провести у 50-70% больных, нормализацию или улучшение зрительных функций отмечают у 80% больных, среди пациентов с предшествующим гипопитуитаризмом нормализацию поврежденных тропных функций гипофиза после аденомэктомии наблюдают до 25% случаев.
Примерно у 12% пациентов с радикальным удалением опухоли отмечаются рецидивы в среднем через 53 мес наблюдения. При динамическом наблюдении среди пациентов с нерадикальным удалением примерно в 60% случаев отмечается увеличение размеров опухоли по данным МРТ, в 17-20% случаев появляются симптомы, связанные с объемом опухоли.
В случае нерадикального оперативного лечения рекомендуют облучение остаточной опухолевой ткани. Сочетание хирургического и лучевого методов лечения обусловливает в последующем прогрессивное снижение активности гипофиза.
Лучевое лечение
Лучевое лечение (как радиотерапевтическое, так и радиохирургическое: см. «Лучевые методы лечения») применяют либо в тех случаях, когда опухоль не удалось удалить радикально, либо как самостоятельный метод лечения, когда другие возможности лечения неприменимы.
В группе больных, которым проводилось облучение остаточной ткани опухоли после нерадикальной аденомэктомии, рецидивы ГНОГ наблюдают в 10-20% случаев, в то время как без лечения дальнейший рост опухоли наблюдают у 50-55% пациентов. Кроме того, рецидив опухоли после нейрохирургического лечения без облучения возникает в среднем через 6 лет, в то время как после комбинированного лечения - через 10 лет.
Как самостоятельный метод лечения облучение применяют при противопоказаниях к хирургическому лечению и при отсутствии значительных неврологических или зрительных нарушений.
После дистанционной телегамма-терапии контроля над размерами опухоли удается достичь в 97-98% случаев в среднем через 57 мес после облучения, однако частота новых случаев гипопитуитаризма может достигать 34-56%.
После радиохирургического лечения с использованием установки «гамма-нож» контроль над размером опухоли достигается у 94 и 85% пациентов через 5 и 10 лет соответственно. Частота новых случаев гипопитуитаризма составляет примерно 24% через 37 мес после облучения.
Медикаментозное лечение
Медикаментозную терапию применяют в тех случаях, когда хирургическое лечение не показано или противопоказано. До сих пор считают, что надежная медикаментозная терапия при ГНОГ отсутствует. В ткани опухолей in vitro были выявлены дофаминовые и соматостатиновые (sst, подтип 2) рецепторы, поэтому в этой группе больных оправдано применение агонистов дофамина и аналогов соматостатина.
Исследования с применением каберголина в дозе 2 мг/нед у больных с ГНОГ в течение 6-12 мес показали, что значимое (на 25% и более) уменьшение размеров опухоли отмечается у 32-56% больных, в то время как только у 20% больных может отмечаться дальнейшее некоторое увеличение размеров опухоли. Поэтому можно считать этот метод медикаментозного лечения достаточно эффективным и экономически оправданным.
Метод ПЭТ в настоящее время позволяет визуализировать рецепторы в ГНОГ in vivo: с помощью введения 123 I-epidepride - D2-дофаминовые рецепторы, с помощью введения 123 I-Tyr-octreotide - sst2-рецепторы. Назначение октреотида (сандостатина♠ ) больным, у которых выявлены sst2-рецепторы, приводило к уменьшению размера опухоли у 97% больных с ГНОГ. Внедрение ПЭТ в широкую клиническую практику позволит в дальнейшем оптимизировать отбор больных для эффективного медикаментозного лечения.
Важный аспект лечения больных с ГНОГ - коррекция гипопитуитаризма, для чего применяют соответствующую гормональную терапию, которая необходима около 60% больных с ГНОГ (см. «Синдром приобретенного гипопитуитаризма у взрослых»).
Показания к консультации других специалистов
Рекомендуют консультации следующих специалистов:
-
офтальмолога (исследование глазного дна, полей зрения, особенно при супраселлярном распространении опухоли);
-
нейрохирурга (особенно при экстраселлярном распространении опухоли и/ или увеличении размеров опухоли при динамическом наблюдении, в случае клинических признаков апоплексии гипофиза);
-
невролога (особенно при распространении опухоли в кавернозные синусы).
Примерные сроки нетрудоспособности
В случае отсутствия неврологических, офтальмологических нарушений, а также при отсутствии симптомов гипопитуитаризма трудоспособность не нарушена.
В других ситуациях сроки нетрудоспособности в каждом конкретном случае определяют с учетом состояния пациента.
Дальнейшее ведение
Цели наблюдения за пациентами с ГНОГ:
Рецидивы ГНОГ отмечают в 3-15% случаев; продолженный рост опухоли после проведенного оперативного лечения (ложные рецидивы) встречается более часто - до 44% случаев. Среднее время возникновения рецидива после оперативного лечения составляет примерно 6 лет, после комбинированного лечения (нейрохирургического и лучевого) - около 10 лет. Таким образом, пациенты с ГНОГ должны находиться под длительным наблюдением.
Обследование пациента включает:
Обследование проводят каждые 6-12 мес (кратность обследования определяет лечащий врач в зависимости от конкретной ситуации).
Информация для пациента (краткие рекомендации)
Опухоли гипофиза - доброкачественные новообразования гипофиза (небольшой железы размерами до 1 см3 , расположенной в основании головного мозга).
В том случае, если опухоль не продуцирует избытка каких-либо гормонов гипофиза, ее называют гормонально-неактивной. При определенных размерах такие опухоли могут вызывать головные боли и быть причиной нарушения зрения, кроме того, опухоль может сдавить нормальную ткань гипофиза и вызвать уменьшение/прекращение продукции достаточного количества гипофизарных гормонов.
Опухоли, размер которых не превышает размеров самого гипофиза, обычно подлежат наблюдению при помощи МРТ. Если размер опухоли больше допустимого и есть гормональные/зрительные нарушения, опухоль подлежит лечению. В первую очередь рассматривают вопрос о возможности проведения оперативного нейрохирургического удаления опухоли. Помимо хирургического, возможно проведение лучевого или медикаментозного лечения. В каждом конкретном случае тактику лечения определяет врач-нейроэндокринолог совместно с офтальмологом, невропатологом, нейрохирургом.
ПРОГНОЗ
ГНОГ - медленно растущие опухоли доброкачественной природы; злокачественные опухоли встречаются в менее чем 1% случаев. В подавляющем большинстве случаев при адекватном наблюдении и проведении своевременного лечения прогноз для пациента благоприятный.
СПИСОК ЛИТЕРАТУРЫ
-
Babu A., Luque R.M., Glick R., Utset M., Fogelfeld L. Variability in quantitative expression of receptors in nonfunctioning pituitary macroadenomas—an opportunity for targeted medical therapy // Endocr. Pract. - 2014 Jan-Feb. - Vol. 20 (1). - P. 15-25.
-
Berkmann S., Schlaffer S., Buchfelder M. Tumor shrinkage after transsphenoidal surgery for nonfunctioning pituitary adenoma // J. Neurosurg. - 2013 Dec. - Vol. 119 (6). - P. 1447-1452.
-
Chen Y., Li Z.F., Zhang F.X., Li J.X., Cai L., Zhuge Q.C., Wu Z.B. Gamma knife surgery for patients with volumetric classification of nonfunctioning pituitary adenomas: a systematic review and meta-analysis // Eur.J. Endocrinol. - 2013 Sep 14. - Vol. 169 (4). - P. 487-495.
-
Cooper O., Melmed S. Subclinical hyperfunctioning pituitary adenomas: the silent tumors // Best Pract. Res. Clin. Endocrinol. Metab. - 2012 Aug. - Vol. 26 (4). - P. 447- 460.
-
Ding D., Starke R.M., Sheehan J.P. Treatment paradigms for pituitary adenomas: defining the roles of radiosurgery and radiation therapy // J. Neurooncol. - 2014 May. - Vol. 117 (3). - P. 445-457.
-
Ezzat S. et al. The prevalence of pituitary adenomas: a systematic review // Cancer. - 2004. - N 101. - P. 613-619.
-
Ezzat S., Asa S.L. Mechanisms of Disease: The Pathogenesis of Pituitary Tumors // Nat. Clin. Pract. Endocrinol. Metab. - 2006. - Vol. 2. - N 4. - P. 220-230.
-
Garcia E.C., Naves L.A., Silva A.O., de Castro L.F., Casulari L.A., Azevedo M.F. Short-term treatment with cabergoline can lead to tumor shrinkage in patients with nonfunctioning pituitary adenomas // Pituitary. - 2013 Jun. - Vol. 16 (2). - P. 189-194.
-
Lake M.G., Krook L.S., Cruz S.V. Pituitary adenomas: an overview // Am.Fam. Physician. - 2013 Sep 1. - Vol. 88 (5). - P. 319-327.
-
Pivonello R., Matrone C., Filippella M., Cavallo L.M., Di Somma C., Cappabianca P., Colao A., Annunziato L., Lombardi G. Dopamine receptor expression and function in clinically nonfunctioning pituitary tumors: comparison with the effectiveness of cabergoline treatment // J. Clin. Endocrinol. Metab. - 2004 Apr. - Vol. 89 (4). - P. 1674-1683.
НЕЙРОЭНДОКРИННЫЕ ОПУХОЛИ
Иловайская И.А.
СИНОНИМЫ
Карциноидные опухоли (это один из наиболее распространенных подвидов НЭО, однако иногда под этим термином ошибочно подразумевают все НЭО), гастроинтерстинальные опухоли, гастроэнтеропанкреатические опухоли, гастро-энтеропанкреатические НЭО.
Устаревшие названия: опухоли клеток APUD-системы, APUDомы.
ОПРЕДЕЛЕНИЕ
НЭО - гетерогенная группа различных по локализации, характеру роста и клинической симптоматике опухолей, происходящих из нейроэндокринных клеток и, соответственно, имеющих сходные цитологические характеристики.
С современных позиций, нейроэндокринными называют клетки, отвечающие следующим критериям (Langley, 1994):
Эти опухоли могут происходить из островковых клеток поджелудочной железы, нейроэндокринных клеток, диффузно локализующихся по всему дыхательному и ЖКТ, а также парафолликулярных клеток щитовидной железы. Опухоли мозговой ткани надпочечников, гипофиза и ОЩЖ имеют сходные морфофункциональные характеристики, однако рассматриваются отдельно.
Основные группы НЭО:
КОД ПО МКБ-10
E34.0 Карциноидный синдром.
E34.1 Другие состояния гиперсекреции интестинальных гормонов.
E34.8 Другие уточненные эндокринные расстройства.
C15-26. Злокачественные новообразования органов пищеварения.
ЭПИДЕМИОЛОГИЯ
Частота встречаемости НЭО, по данным современных эпидемиологических исследований, составляет примерно 35 случаев на 100 000 населения, частота новых случаев - 2-5 на 100 000 человек в год, карциноидные опухоли составляют примерно 2 /3 случаев, наиболее частая локализация - ЖКТ (70%; в первую очередь - аппендикс и подвздошная кишка) и бронхо-легочная система (25%). Соотношение мужчин и женщин равно 0,86.
Новые случаи с яркой клинической симптоматикой карциноидного синдрома регистрируют с частотой примерно 2 случая на 100 000 населения в год.
Панкреатические нейроэндокринные опухоли составляют примерно 1 /3 случаев от всех НЭО (табл. 14.16).
| Тип опухоли | Частота встречаемости на 1 млн населения | Основная локализация | М: Ж |
|---|---|---|---|
Инсулиномы |
0,8-2,0 |
β-Клетки поджелудочной железы (99%) |
2:3 |
Гастриномы |
0,5-1,5 |
Двенадцатиперстная кишка (70%), головка поджелудочной железы (25%) |
Чаще у мужчин |
Випомы |
0,05-0,2 |
Поджелудочная железа (70-80%) |
1:3 |
Глюкагономы |
0,01-0,1 |
α-Клетки поджелудочной железы (хвост 50-80%, головка 22%, тело 14%) |
Нет данных |
Новые случаи панкреатических эндокринных опухолей - один случай на 100 000 населения в год. Данные аутопсийных исследований свидетельствуют о гораздо большей распространенности панкреатических эндокринных опухолей: при выборочном исследовании поджелудочной железы опухолевые очаги были обнаружены в 0,3-1,6% случаев; в тех исследованиях, где все поджелудочные железы подвергали изучению, частота обнаружения очагов составила до 10% случаев.
Другие НЭО встречаются гораздо реже.
ПРОФИЛАКТИКА
Профилактика не разработана.
СКРИНИНГ
Поскольку НЭО входят в состав синдрома МЭН-1, лиц с верифицированным синдромом МЭН-1, а также их родственников первой линии подвергают скрининговому обследованию, включающему ежегодное определение:
КЛАССИФИКАЦИЯ
Ни одна из предложенных классификаций в настоящее время не является универсальной.
По особенностям происхождения и клинической симптоматике условно выделяют:
-
-
функционально-активные (70-85% случаев):
-
гастринома (продуцирует избыток гастрина, клинически проявляется синдромом Золлингера-Эллисона);
-
ВИПома (продуцирует избыток вазоактивного интестинального полипептида);
-
КРГома (продуцирует избыток кортикотропин-рилизинг гормона, клиническая картина гиперкортицизма);
-
соматолиберинома (опухоль, продуцирующая избыток соматолиберина, - клиническая картина акромегалии);
-
АКТГома (продуцирует избыток АКТГ, характерны АКТГ-эктопированный синдром, клиническая картина эндогенного гиперкортицизма).
Существует классификация, предложенная E.D. Williams в 1963 г., в которой карциноидные НЭО подразделяются по отделу эмбриональной кишечной трубки, из которого возникла первичная опухоль, так как в зависимости от происхождения нейроэндокринных клеток клинико-биохимические свойства опухолей могут различаться (табл. 14.17).
| Отдел эмбриональной кишечной трубки | Группа опухолей | Характеристика |
|---|---|---|
Верхний (foregut) |
Опухоли тимуса, легких, желудка, двенадцатиперстной кишки и поджелудочной железы |
Низкое содержание серотонина, высокое содержание предшественника серотонина - 5-гидрокситриптофана, усиленная секреция гистамина, атипичное течение карциноидного синдрома. Высокий риск метастазирования в кости |
Средний (midgut) |
Опухоли тонкой кишки, аппендикса, правых отделов толстой кишки |
Редкая гормональная секреция. Усиленная секреция серотонина и вазоактивных субстанций. Высокий риск метастазирования в печень |
Концевой (hindgut) |
Левые отделы ободочной кишки, прямая кишка |
Так как принципиально важным является морфологическое строение НЭО, значительное внимание уделяется морфологическим классификациям НЭО, которые постоянно совершенствуются. Последний пересмотр НЭО ЖКТ был в 2010 г., а в классификацию НЭО тимуса и легких были внесены последние изменения в 2015 г. Добавлен индекс пролиферации Ki-67 как фактор градации. Однако для НЭО ЖКТ используется термин «НЭО», а для легких и тимуса - «карциноид». Но, несмотря на различия в терминологии и вариации критериев принадлежности опухоли к той или иной категории, каждая классификация выделяет 3 степени злокачественности НЭО (табл. 14.18).
| Степень злокачественности, Grade | Легкие и тимус, ВОЗ, 2015 | НЭО ЖКТ, ВОЗ, 2010 |
|---|---|---|
Низкая степень злокачественности (low grade) |
Типичный карциноид |
НЭО, G1 |
Средняя степень злокачественности (intermediate grade) |
Атипичный карциноид |
НЭО, G2 |
Высокая степень злокачественности (high grade) |
Мелкоклеточная нейроэндокринная карцинома |
НЭО, G3, мелкоклеточная карцинома |
Крупноклеточная нейроэндокринная карцинома |
НЭО, G3, крупноклеточная нейроэндокринная карцинома |
Степень злокачественности опухоли с указанием классификации, к которой она относится, должна обязательно включаться в патоморфологическое заключение.
Степень злокачественности НЭО определяется на основании митотической и пролиферативной активности опухолевых клеток, определяемой при патоморфологическом исследовании (табл. 14.19).
| Степень злокачественности, Grade | Легкие и тимус, ВОЗ, 2015 | НЭО ЖКТ, ВОЗ, 2010 |
|---|---|---|
Низкая степень злокачественности (low grade) |
<0-1 митоз / 10 репрезентативных полей зрения* <5% индекс Ki-67 Отсутствие некрозов |
<2 митозов / 10 репрезентативных полей зрения <3% индекс Ki67** |
Средняя степень злокачественности (intermediate grade) |
2-10 митозов / 10 репрезентативных полей зрения <20% индекс Ki-67 Нет или фокусы некроза |
2-20 митозов / 10 репрезентативных полей зрения или 3-20% индекс Ki67 |
Высокая степень злокачественности (high grade) |
>10 митозов / 10 репрезентативных полей зрения 40-80% индекс Ki-67 (крупноклеточная нейроэндокринная карцинома) 50-100% индекс Ki-67 (для мелкоклеточной нейроэндокринной карциномы) Обширные поля некрозов |
>20 митозов / 10 репрезентативных полей зрения или >20% индекс Ki67 |
* Подсчет митотического индекса производится в препаратах, окрашенных гематоксилином и эозином в 50 репрезентативных полях зрения при большом увеличении, а митотический индекс выражается как среднее в 10 репрезентативных полях зрения.
** Для определения индекса Ki-67 следует использовать клон MIB-1 и вычислять как среднее число меченых ядер на 100 учтенных (%) при подсчете оптимально 2000 опухолевых клеток в репрезентативных полях зрения и с учетом участков с наиболее высокой ядерной экспрессией.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Основная причина возникновения НЭО - генетические мутации. Известно достаточное количество генов, вовлеченных в туморогенез, - MEN1, RET, VHL, TSC1, TSC2 и другие. Мутации MEN1 - наиболее распространенная причина возникновения НЭО. При синдроме МЭН-1 в большинстве случаев обнаруживают герминальные мутации (95%), однако могут быть и спорадические мутации (более подробно см. «МЭН-1»).
Каскад генетических изменений приводит к активации онкогенов и/или инактивации туморсупрессивных генов, в результате чего запускаются процессы клеточной трансформации и пролиферации. Сначала возникает гиперплазия клеток, затем формируются диспластичные клетки, которые под влиянием различных ростовых факторов (фактора роста опухоли, фактора роста фибробластов β, сосудистого ростового фактора, фактора некроза опухоли и др.) преобразуются в высокодифференцированные опухоли. Дальнейшая активация онкогенов (например, c-myc, K-ras) может приводить к дедифференцировке клеток, формированию умеренно-дифференцированной опухоли. Потеря следующего пула генов - туморсупрессорных (например, PTEN) и контролирующих апоптоз, а также хромосомная нестабильность приводят к развитию низкодифференцированных опухолей с высокой способностью к метастазированию.
Карциноидные опухоли, которые развиваются из энтерохромаффинных клеток (или клеток Кульчицкого), и инсулиномы, которые развиваются из клеток островков Лангерганса, в 80-90% случаев доброкачественные, так как в процессы трансформации вовлекаются зрелые дифференцированные клетки. Источником других НЭО (таких, как гастринома, випома, соматостатинома и др.) служит мультипотентная стволовая клетка, дающая начало как эндокринным, так и экзокринным клеткам. Потенциальные возможности злокачественного роста этих новообразований обнаруживают в 60-70% случаев.
Так называемые нефункционирующие НЭО сохраняют способность к синтезу и секреции гормонов, среди которых наиболее часто отмечают хромогранины А (до 90% случаев) и панкреатический полипептид (до 70%).
Карциноидные опухоли часто сопровождаются поражением сердечнососудистой системы. Кардиотоксичные вещества, вырабатываемые опухолью, инактивируются в печени и легких, поэтому при карциноиде ЖКТ поражаются только правые отделы сердца, и лишь при наличии метастазов в печень. Левые отделы сердца страдают при карциноиде ЖКТ только когда есть сброс крови справа налево, а также при карциноиде бронхов. На эндокарде, клапанах и в крупных сосудах, вероятно вследствие травмы эндотелия, появляются фиброзные бляшки. Они состоят из гладкомышечных клеток, окруженных коллагеном и кислыми гликозаминогликанами. Типичные клапанные пороки - трикуспидальная недостаточность, стеноз клапана легочной артерии и их сочетание. Если опухоль вырабатывает сосудорасширяющие вещества, то возможно падение общего периферического сосудистого сопротивления и повышение сердечного выброса, а если сосудосуживающие - спазм коронарных артерий.
Панкреатические эндокринные опухоли вырабатывают вещества, в той или иной степени контролирующие обмен веществ и деятельность ЖКТ, поэтому один из наиболее распространенных симптомов - нарушение моторики ЖКТ.
КЛИНИЧЕСКАЯ КАРТИНА
Клиническая картина заболевания зависит от спектра гормональной активности, которой обладает НЭО.
Карциноидный синдром. Классическую клиническую картину наблюдают у 10% пациентов с карциноидным синдромом. Она включает следующие симптомы:
-
Все проявления непосредственно связаны с повышением содержания серотонина, которое обнаруживают у 93-94% пациентов с данной симптоматикой. У пациентов с карциноидными опухолями без классической клинической картины повышение содержания серотонина отмечают в 25-30% случаев.
-
К моменту постановки первичного диагноза метастазы обнаруживают у 45% больных. Чаще всего метастазирование поражает поджелудочную железу (76% случаев), тонкую и толстую кишку (71%).
Инсулиномы. Симптоматика связана с основным биохимическим проявлением гиперсекреции инсулина - гипогликемией. Клиническая картина включает симптомы снижения содержания глюкозы и активации адренергической системы, а также нейрогликопенические симптомы, связанные с хроническим отсутствием достаточного количества глюкозы в нервной ткани:
Гастриномы (синдром Золлингера-Эллисона). Основные симптомы:
-
В момент диагностики заболевания метастазы в печень и лимфатические узлы обнаруживают в 75-80% случаев, метастазы в кости - в 12% случаев. Гастриномы обычно маленького размера, размер коррелирует с метастазированием в печень: менее 1 см - метастазы у 4% пациентов, 1-2,9 см - в 28% случаев; свыше 3 см - 61%.
Випомы (синдром Вернера-Моррисона).
Глюкагономы .
ДИАГНОСТИКА
Анамнез и физикальное обследование
При сборе анамнеза необходимо уточнить длительность заболевания. Часто при осмотре отмечают снижение массы тела.
Можно обнаружить изменения кожных покровов и слизистых оболочек:
-
при карциноидном синдроме во время прилива - покраснение лица, верхней части туловища по сосковым линиям, наличие телеангиэктазий;
-
при атипичном течении карциноидного синдрома кожа может приобретать пурпурно-фиолетовый цвет, на месте эритемы далее образуются множественные телеангиэктазии;
-
при глюкагономах - некротизирующий дерматит, стоматит. Почти при всех НЭО отмечают артериальную гипотензию.
Лабораторные исследования
В соответствии с рекомендациями Европейского общества по НЭО, хромогранин А является обязательным маркером биохимического обследования в целях диагностики, мониторинга и прогноза НЭО. Хромогранин А характеризуется высокой диагностической чувствительностью при НЭО желудка (95%), подвздошной кишки (80%), синдроме МЭН (78%), бронхолегочной системы (70%). В различных исследованиях была выявлена зависимость секреции хромогранина А от распространенности НЭО.
-
-
Определение общего белка в крови (характерна белковая недостаточность, так как 50% пищевого триптофана расходуется на синтез серотонина).
-
Определение концентрации серотонина и его основного метаболита - 5-гидроксииндолуксусной кислоты в суточной моче (экскреция более 150 мкмоль за 24 ч свидетельствует о наличии карциноидного синдрома).
-
Инструментальные исследования
При подозрении на тот или иной вид НЭО обычно необходимо проведение:
-
эндоУЗИ ЖКТ с визуализацией поджелудочной железы (особенно чувствительный метод диагностики поражения поджелудочной железы);
-
МРТ органов брюшной полости (имеет преимущества по сравнению с КТ);
-
сканирования с меченым октреотидом (обнаружение соматостатиновых рецепторов в опухолях);
-
ПЭТ-КТ или ПЭТ-МРТ с различными конрастными веществами в зависимости от вида опухоли (например, при диагностике карциноидных опухолей используют предшественник серотонина 11-С-5-НТР, который накапливается в первичном очаге, а также в метастазах).
ПОКАЗАНИЯ К КОНСУЛЬТАЦИИ ДРУГИХ СПЕЦИАЛИСТОВ
ПРИМЕР ФОРМУЛИРОВКИ ДИАГНОЗА
Карциноидный синдром тяжелого течения. НЭО двенадцатиперстной кишки. Синдром Золлингера-Эллисона. Опухоль поджелудочной железы (гастринома). Метастатическое поражение левой доли печени.
ЛЕЧЕНИЕ
Цели лечения
Показания к госпитализации
Немедикаментозное лечение
Для лечения НЭО используют современные немедикаментозные методы терапии.
-
Эмболизация печеночной артерии (поливиниловыми алкогольсодержащими микросферами) - эффективна при метастазах в печень, улучшает состояние 40-90% пациентов на средний период 10-15 мес. Метод непростой, существуют протоколы ведения больных, необходима определенная медикаментозная подготовка к процедуре. Побочные эффекты (до 10% случаев) включают печеночные абсцессы, почечную недостаточность, некрозы желчного пузыря и тонкой кишки, однако если у конкретного пациента достигнут положительный эффект, процедуру можно повторить при ухудшении состояния.
-
Печеночная химиоэмболизация - окклюзия печеночной артерии с проведением локальной цитотоксической химиотерапии. Метод паллиативный, однако дает положительный результат у 63% пациентов, улучшает состояние в среднем на два года.
-
Метод печеночной радиоэмболизации, когда для окклюзии печеночной артерии вводят микросферы, содержащие радиоизотопы, паллиативный метод лечения, однако, облегчает симптомы и замедляет рост опухоли.
-
Метод радионуклидной терапии пептидных рецепторов с использованием радиомеченных аналогов соматостатина. Этот метод можно применять только при НЭО с обнаруженными при сканировании соматостатиновыми рецепторами. Применяется у пациентов с высокой распространенностью процесса, не подлежащих хирургическому лечению, стабилизация заболевания достигается после процедуры у 35% больных, регресс опухоли - у 10%.
Медикаментозное лечение
В лечении НЭО используют медикаментозную терапию интерфероном, аналогами соматостатина и химиотерапию, в том числе ингибитор mTOR эверолимус и ингибитор тирозинкиназ сунитиниб. Выбор тактики лечения зависит от вида и характеристик НЭО.
-
Интерферон используют в дозах 3-9 МЕ три раза в неделю, применение более высоких доз не дает повышения эффективности препарата. Согласно анализу сводных данных об эффективности интерферона в лечении больных с НЭО, среди больных с карциноидными опухолями улучшение биохимических показателей отмечают в 42% случаев, регресс опухоли - в 14% случаев; среди больных с панкреатическими НЭО биохимическое улучшение достигают в 51% случаев, регресс опухоли - в 12%. Лечение интерфероном показано при опухолях с низкой пролиферативной активностью как лечение первой или второй линии после химиотерапии. Побочные эффекты возникают достаточно часто: лихорадочное состояние (89%), потеря массы тела (59%), слабость (51%), депрессия (50%). Существует также риск развития гепатотоксичности, аутоиммунных заболеваний и появления нейтрализующих антител. При клинически манифестированных НЭО интерферон используют совместно с аналогами соматостатина, хотя проведенные рандомизированные исследования не подтвердили повышение эффективности лечения сочетанием этих двух препаратов по сравнению с монотерапией.
-
Аналоги соматостатина (пролонгированные формы) успешно применяют в лечении НЭО, так как соматостатиновые рецепторы представлены как в первичных опухолях, так и в их метастазах. Применение октреотида значительно улучшает состояние больного, способствуя существенному снижению гормональной секреции опухоли: при випомах - в 89% случаев, при глюкагономах - до 75% случаев, при инсулиномах - до 50-60% случаев. Клинически это проявляется в уменьшении или прекращении диареи у 40-60% больных, что приводит к улучшению качества жизни. На фоне применения аналогов соматостатина отмечается стабилизация размеров опухоли у 50-70%, в крупном исследовании PROMID было показано увеличение среднего времени до прогрессии опухоли с 2-5 мес до 14,3 мес, а также увеличение общей выживаемости на фоне лечения аналогами соматостатина. В 2013 г. были представлены данные исследования II фазы, которое позволяет предположить повышение эффективности терапии аналогами соматоста-тина после добавления эверолимуса, среднее время до прогрессии опухоли увеличивалось до 16,3 мес.
-
Химиотерапия показана в первую очередь для лечения низкодифференцированных быстрорастущих (более 25% от исходного объема) метастатических опухолей, биологический ответ отмечают в 20-69% случаев. В лечении высокодифференцированных (особенно панкреатических) НЭО, обладающих медленными темпами роста, химиотерапия имеет ограниченное значение, ее применяют в случае отсутствия эффекта от других видов лечения. В исследовании III фазы с целью оценки эффективности сунитиниба в лечении некурабельных НЭО было отмечено увеличение периода до прогресии опухоли с 5,5 до 11,4 мес по сравнению с плацебо. Химиотерапевтические препараты, используемые в лечении НЭО, представлены в табл. 14.20.
| Вид терапии | Вид опухоли |
|---|---|
Стрептозоцин + доксорубицин |
Высокодифференцированные панкреатические НЭО |
Ломустин, стрептозоцин + фторурацил (5-фторурацил♠) |
Генерализованные НЭО |
Этопозид + цисплатин |
Низкодифференцированные, агрессивные НЭО, панкреатические опухоли, карциноидные опухоли (foregut) |
Сунитиниб |
Нерезектабельные или метастатические, высокодифференцированные НЭО поджелудочной железы у взрослых с прогрессированием заболевания |
Эверолимус |
Распространенные и/или метастатические НЭО ЖКТ, легкого и поджелудочной железы |
Симптоматическая терапия также зависит от вида НЭО (табл. 14.21). При карциноидных опухолях используют блокаторы серотониновых/гистаминовых рецепторов, однако применение их дает симптоматическое улучшение, но не предупреждает и не останавливает развитие изменений в сердце.
| Вид опухоли | Вид терапии |
|---|---|
Карциноидные опухоли |
Аналоги соматостатина, если результаты сканирования с октреотидом положительные. Интерферон, если рецепторы к соматостатину не обнаружены. Антагонисты гистамина. Ципрогептадин, никотинамид |
Инсулинома |
Диазоксид. Частое дробное углеводистое питание. Внутривенное введение раствора декстрозы при отсутствии возможности перорального питания. Аналоги соматостатина, если результаты сканирования с октреотидом положительные |
Гастринома |
Ингибиторы протоновой помпы в высоких дозах |
Глюкагонома |
Высокие дозы аналогов соматостатина. Антикоагулянтная терапия, так как есть проявления тромбофилии. Инсулинотерапия для коррекции диабета |
Випома |
Высокие дозы аналогов соматостатина. Массивная интравенозная регидратация для ликвидации обезвоживания. Компенсация солевых потерь (калий, бикарбонаты) |
Соматостатинома |
Панкреатические ферменты. Инсулинотерапия для коррекции диабета |
Нефункционирующие опухоли |
Аналоги соматостатина, если результаты сканирования с октреотидом положительные |
Хирургическое лечение
Хирургическое лечение - оптимальный и наиболее эффективный метод терапии. По данным Национального института рака (США), в группе больных, радикально прооперированных по поводу НЭО, десятилетняя выживаемость составила 94%, метастатическое поражение печени было отмечено лишь в 3% случаев. Однако в 65-70% случаев НЭО носит злокачественный характер, и на момент первичного обращения к врачу уже есть метастазы в печень и другие органы.
Кроме того, проводят паллиативное хирургическое лечение (удаление части опухоли) с целью ликвидации патологического сдавления опухолью окружающих органов и тканей, пятилетняя выживаемость пациентов после паллиативной резекции опухоли составляет 63%.
Если возможно, проводят хирургическое удаление метастазов в печень и лимфатические узлы. При поражении печени один из методов лечения - трансплантация печени, через год выживаемость составляет 53%. Применяют также метод криохирургии, если поражено менее 40% печени: в каждый метастатический очаг вводят криопробы, вызывающие локальный некроз опухоли. В большинстве случаев лечение метастатического процесса при НЭО невозможно без дополнительного медикаментозного лечения.
Применяют и симптоматическое хирургическое лечение - при карциноидном синдроме в тяжелых случаях показано протезирование сердечных клапанов.
Показания к консультации других специалистов
Ведение пациентов осуществляют эндокринологи и хирурги совместно с онкологами, при необходимости - с радиологами и химиотерапевтами.
Примерные сроки нетрудоспособности
Пациенты с клиническими проявлениями НЭО в подавляющем большинстве случаев нетрудоспособны. Восстановление трудоспособности зависит от характеристик НЭО, доброкачественности или злокачественности распространения опухоли, радикальности выполнения хирургической операции и т.д.
Дальнейшее ведение
После радикально выполненного лечения (если это возможно) пациенты должны находиться под динамическим наблюдением, необходимо проводить регулярное (один раз в 3-6-12 мес) обследование с целью исключения рецидивов и метастазов (гормональное обследование, УЗИ печени, по показаниям - КТ/МРТ органов брюшной полости).
ПРОГНОЗ
При обнаружении карциноидной опухоли до появления клинической картины карциноидного синдрома пятилетняя выживаемость составляет в целом 50%, после появления клинической симптоматики - 30-47% (появление симптомов часто служит признаком более поздней стадии заболевания). Более подробно данные представлены в табл. 14.22 и 14.23.
Локализация опухоли |
% от всех карциноидов |
Частота метастазов, % |
|
регионарных |
отдаленных |
||
Легкие |
25,3 |
5,2 |
0,5 |
Тонкая кишка |
28,2 |
35,9 |
22,4 |
Аппендикс |
2,4 |
28,9 |
9,9 |
Толстая кишка |
7,6 |
25,8 |
29,5 |
Прямая кишка |
18,5 |
2,2 |
1,7 |
Желудок |
5,9 |
3,1 |
6,5 |
Яичник |
1,4 |
5,2 |
0,5 |
Локализация опухоли |
Пятилетняя выживаемость, % |
||
без метастазов |
с метастазами |
||
регионарными |
отдаленными |
||
Легкие |
81,1 |
76,7 |
25,6 |
Тонкая кишка |
59,9 |
72,8 |
50 |
Аппендикс |
80,8 |
88,1 |
9,6 |
Толстая кишка |
76 |
71,6 |
30 |
Прямая кишка |
90,8 |
48,9 |
32,3 |
Желудок |
69,1 |
Нет данных |
21,2 |
Яичник |
90,9 |
Нет данных |
28,3 |
При хирургическом удалении гастриномы без метастазов в печени пятилетняя выживаемость составляет 50%, при метастазах в печень - 30%.
СПИСОК ЛИТЕРАТУРЫ
-
Орлова Р.В., Новик А.В. Современные подходы лекарственного лечения генерализованных форм нейроэндокринных опухолей. Симптоматическая терапия синдромов при нейроэндокринных неоплазиях // Практическая онкология. - 2005. - Т. 6, №4. - С. 240-246.
-
Artale S., Giannetta L., Cerea G. Treatment of metastatic neuroendocrine carcinomas based on WHO classification // Anticancer Res. - 2005. - Vol. 25. - P. 4463-4469.
-
Bison S.M., Konijnenberg M.W., Melis M., Pool S.E., Bernsen M.R., Teunissen J.J., Kwekkeboom D.J., de Jong M. Peptide receptor radionuclide therapy using radiolabeled somatostatin analogs: focus on future developments // Clin. Transl. Imaging. - 2014. - Vol. 2. - P. 55-66.
-
Cives M., Strosberg J. An Update on Gastroenteropancreatic Neuroendocrine Tumors // Oncology (Williston Park). - 2014 Sep 15. - Vol. 28 (9).
-
Hemminki K., Li X. Incidence trends and risk factors of carcinoid tumors: a nationwide epidemiologic study from Sweden // Cancer. - 2001. - Vol. 92. - P. 2204-2210.
-
Karakaxas D., Gazouli M., Liakakos T., Vaiopoulou A., Apessou D., Papaparaskeva K., Patapis P., Dervenis C. Pancreatic neuroendocrine tumors: current opinions on a rare, but potentially curable neoplasm // Eur.J. Gastroenterol. Hepatol. - 2014 Aug. - Vol. 26 (8). - P. 826-835.
-
Karampelas I. N., Syrigos K. N., Saif M. W. Targeted agents in treatment of neuroendocrine tumors of pancreas // JOP. - 2014 Jul 28. - Vol. 15 (4). - P. 351-353.
-
Klimstra D.S., Modlin I.R., Coppola D., Lloyd R.V., Suster S. The Pathologic Classification of Neuroendocrine Tumors. A review of Nomenclature, Grading, and Staging Systems // Pancreas. - 2010. - Vol. 39. - P. 707-712.
-
Strosberg J.R., Benson A.B., Huynh L., Duh M.S., Goldman J., Sahai V., Rademaker A.W., Kulke M.H. Clinical Benefits of Above-Standard Dose of Octreotide LAR in Patients With Neuroendocrine Tumors for Control of Carcinoid Syndrome Symptoms: A Multicenter Retrospective Chart Review Study // Oncologist. - 2014 Sep. - Vol. 19 (9). - P. 930- 936.
-
Triavis W.D., Brambilla E., Burke A.P., Marx A., Nicholson AG. WHO classification of tumours of the Lung, Pleura, Thymus and Heart. - Lyon: IARC; 2015.
НЕСАХАРНЫЙ ДИАБЕТ
Дзеранова Л.К., Пигарова Е.А.
СИНОНИМЫ
Гипоталамический несахарный диабет, гипофизарный несахарный диабет, нейрогипофизарный несахарный диабет, несахарное мочеизнурение.
ОПРЕДЕЛЕНИЕ
Несахарный диабет - заболевание, характеризующееся неспособностью почек реабсорбировать воду и концентрировать мочу, имеющее в своей основе дефект секреции или действия вазопрессина и проявляющееся выраженной жаждой и экскрецией большого количества разведенной мочи.
КОД ПО МКБ-10
ЭПИДЕМИОЛОГИЯ
Распространенность несахарного диабета в популяции по различным источникам составляет 0,004-0,01%.
ПРОФИЛАКТИКА
Профилактика не разработана.
СКРИНИНГ
Скрининг не проводят.
КЛАССИФИКАЦИЯ
В клинической практике различают три основных типа несахарного диабета:
-
центральный (гипоталамический, гипофизарный), обусловленный нарушением синтеза или секреции вазопрессина;
-
нефрогенный (почечный, вазопрессин-резистентный), который характеризуется резистентностью почек к действию вазопрессина;
-
первичная полидипсия: нарушение, когда патологическая жажда (дипсогенная полидипсия) или компульсивное желание пить (психогенная полидипсия) и связанное с этим избыточное потребление воды подавляют физиологическую секрецию вазопрессина, в итоге приводя к характерной симптоматике несахарного диабета, при этом при дегидратации организма синтез вазопрессина восстанавливается.
Также выделены и другие, более редкие типы несахарного диабета:
-
гестагенный, связанный с повышенной активностью фермента плаценты - аргининаминопептидазы, разрушающей вазопрессин;
-
функциональный: возникает у детей первого года жизни и обусловлен незрелостью концентрационного механизма почек и повышенной активностью фосфодиэстеразы 5-го типа, что приводит к быстрой деактивации рецептора к вазопрессину и низкой продолжительности действия гормона;
-
ятрогенный: к этому типу относят применение диуретиков, выполнение рекомендаций потребления больших объемов жидкости.
По тяжести течения:
По степени компенсации:
-
компенсация - при лечении жажда и полиурия в целом не беспокоят;
-
субкомпенсация - при лечении бывают эпизоды жажды и полиурии в течение дня, оказывающие влияние на повседневную деятельность;
-
декомпенсация - жажда и полиурия сохраняются и при лечении заболевания и оказывают существенное влияние на повседневную деятельность.
ЭТИОЛОГИЯ
ПАТОГЕНЕЗ
Патогенез центрального несахарного диабета: нарушение секреции или действия вазопрессина на V2-рецептор (рецептор к вазопрессину 2-го типа) главных клеток собирательных трубочек приводит к тому, что не происходит «встраивания» вазопрессин-чувствительных водных каналов (аквапоринов 2) в апикальную клеточную мембрану, и, следовательно, нет реабсорбции воды. При этом вода в большом количестве теряется с мочой, вызывая обезвоживание и, как следствие, жажду.
КЛИНИЧЕСКАЯ КАРТИНА
Основные проявления несахарного диабета - выраженная полиурия (выделение мочи более 2 л/м2 в сутки или 40 мл/кг в сутки у старших детей и взрослых), полидипсия (порядка 3-18 л/сут) и связанные с ними нарушения сна. Характерно предпочтение простой холодной/ледяной воды. Могут быть сухость кожи и слизистых оболочек, уменьшение слюно- и потоотделения. Аппетит, как правило, снижен. Систолическое АД может быть нормальным или немного пониженным при характерном повышении диастолического АД. Тяжесть заболевания, т.е. выраженность симптомов, зависит от степени нейросекреторной недостаточности. При частичном дефиците вазопрессина клиническая симптоматика может быть не столь отчетлива и проявляться только в условиях питьевой депривации или избыточной потери жидкости (походы, экскурсии, жаркая погода). В связи с тем, что глюкокортикоиды необходимы почкам для выделения воды, не содержащей электролитов, симптомы центрального несахарного диабета могут маскироваться сопутствующей надпочечниковой недостаточностью, и в таком случае назначение терапии глюкокортикоидами приводит к манифестации/усилению полиурии.
ДИАГНОСТИКА
Анамнез
При сборе анамнеза необходимо уточнять длительность и стойкость симптомов у пациентов, наличие полидипсии, полиурии, ранее выявленных нарушений углеводного обмена и наличие СД у родственников.
Физикальное обследование
При осмотре могут быть обнаружены симптомы дегидратации: сухость кожи и слизистых оболочек. Систолическое АД нормальное или немного пониженное, диастолическое АД повышено.
Лабораторные исследования
Для несахарного диабета характерно увеличение осмоляльности крови, гипернатриемия, постоянно низкие осмоляльность (<300 мОсм/кг) или относительная плотность мочи (<1005 г/л). Для первичной полидипсии - снижение осмоляльности крови и гипонатриемия на фоне такой же низкой осмоляльности и относительной плотности мочи. Необходимо проведение клинического анализа мочи, а также определение концентрации калия, кальция, глюкозы, мочевины и креатинина в биохимическом анализе крови для исключения воспалительных заболеваний почек и наиболее частых электролитно-метаболических причин возникновения нефрогенного несахарного диабета.
Показано генетическое исследование при подозрении на наследственный характер заболевания.
Инструментальные исследования
При нефрогенном несахарном диабете:
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Точная дифференциальная диагностика основных трех форм несахарного диабета принципиальна для выбора лечения, а также дальнейшего поиска возможной причины заболевания и патогенетического лечения. В ее основе лежат три этапа.
-
На первом этапе подтверждают наличие гипотонической полиурии - выделение мочи более 2 л/м2 в сутки или 40 мл/кг в сутки у старших детей и взрослых с относительной плотностью менее 1005 г/л или осмоляльностью менее 300 мОсм/кг.
-
На втором этапе проводят пробу с сухоедением (исключение первичной полидипсии) и десмопрессиновый тест (для разделения центрального и нефрогенного типов несахарного диабета).
Протокол классической пробы с сухоедением (дегидратационный тест) по G.L. Robertson (для подтверждения несахарного диабета) Исходные действия:
В дальнейшем через равные промежутки времени в зависимости от состояния больного через 1 или 2 ч повторять эти действия.
Во время пробы: больному не разрешается пить, желательно также ограничение пищи (по крайней мере, в течение первых 8 ч проведения пробы); при кормлении пища не должна содержать много воды и легкоусвояемых углеводов, можно вареные яйца, зерновой хлеб, нежирные сорта мяса, рыбы.
Пробу прекращают при:
Проведение пробы с сухоедением в амбулаторных условиях
Только для пациентов в стабильном состоянии, с подозрением на наличие полидипсии и выделяющих до 6-8 л/сут. Цель - получение наиболее концентрированной (последней) порции мочи.
Методика проведения
-
Попросить больного полностью ограничить прием жидкости в течение того времени, которое он сможет выдержать. Наиболее удобно начало ограничения за несколько часов до сна и во время ночного сна.
-
Пациент собирает пробы мочи при возникновении естественной необходимости мочеиспускания ночью и при пробуждении, при этом на анализ приносится только самая последняя порция, поскольку она будет в условиях полного ограничения жидкости являться самой концентрированной.
-
До проведения анализа мочу хранят в закрытом виде в холодильнике.
-
Пробу может прекратить сам пациент, руководствуясь своим самочувствием, тогда для анализа он приносит самую последнюю порцию мочи до возобновления питья.
-
В последней порции мочи определяется осмоляльность/осмолярность: показатель, превышающий 650 мОсм/кг, позволяет исключить любой генез несахарного диабета.
Проведение десмопрессинового теста (для дифференциальной диагностики центральной и нефрогенной форм несахарного диабета) по G.L. Robertson
Проводится пациентам после исключения полидипсии, оптимально после пробы с сухоедением. Методика проведения:
-
ввести 2 мкг десмопрессина в/в, в/м или п/к, или 10 мкг интраназально, или 0,1 мг таблетированного десмопрессина под язык до полного рассасывания, или 120 мкг десмопрессина в виде таблеток подъязычных;
-
пациенту разрешается есть и пить (объем выпиваемой жидкости не должен превышать объема выделенной мочи во время фазы дегидратации);
-
через 2 и 4 ч собрать мочу для определения объема и осмоляльности;
-
на следующее утро взять кровь для определения натрия и осмоляльности, собрать мочу для определения объема и осмоляльности.
У большинства пациентов функциональное состояние центра жажды полностью сохранено, в связи с чем нормонатриемия и нормальная осмоляльность крови поддерживаются путем потребления жидкости, адекватной потерям. Биохимические изменения становятся очевидными только при ограничении доступа больных к воде и при патологии центра жажды. Таким пациентам для подтверждения диагноза «несахарный диабет» (т.е. исключения психогенной и дипсогенной полидипсии) необходимо проведение пробы с сухоедением. Во время дегидратации, несмотря на уменьшение объема циркулирующей крови, снижение клубочковой фильтрации и повышение осмоляльности и натрия крови, полиурия сохраняется, концентрация мочи и ее осмоляльность почти не возрастают (относительная плотность мочи 1000-1005 г/л, осмоляльность мочи ниже осмоляльности плазмы, т.е. ниже 300 мОсм/кг). Это приводит к развитию симптомов обезвоживания: сухости губ и слизистой рта, резкой общей слабости, тахикардии, гипотензии, коллапсу. По мере нарастания дегидратации организма появляются также головная боль, тошнота, рвота, которая усугубляет дефицит жидкости и электролитов, лихорадка, сгущение крови с повышением концентрации натрия, гемоглобина, остаточного азота, количества эритроцитов. Возникают судороги, психомоторное возбуждение.
ПОКАЗАНИЯ К КОНСУЛЬТАЦИЯМ СПЕЦИАЛИСТОВ
При подозрении на наличие патологических изменений гипоталамо-гипофизарной области показаны консультации нейрохирурга и офтальмолога; при обнаружении патологии мочевыводящей системы - уролога, а при подтверждении психогенного варианта полидипсии необходимо направление на консультацию к психиатру/психоневрологу. При подозрении на развитие центрального несахарного диабета в рамках DIDMOAD-синдрома проводится дополнительное обследование на наличие СД, обследование у офтальмолога для исключения атрофии зрительных нервов, ЛОРа - нейросенсорной тугоухости.
ПРИМЕР ФОРМУЛИРОВКИ ДИАГНОЗА
Центральный несахарный диабет средней степени тяжести, медикаментозная компенсация.
ЛЕЧЕНИЕ
При подтвержденном несахарном диабете необходимо установить свободный (в соответствии с потребностью/жаждой) питьевой режим.
При центральном несахарном диабете назначают синтетический аналог вазопрессина - десмопрессин. Десмопрессин активирует только V2-рецепторы вазопрессина главных клеток собирательных канальцев почек. По сравнению с вазопрессином десмопрессин обладает менее выраженным действием на гладкие мышцы сосудов и внутренних органов, обладая большей антидиуретической активностью, а также более устойчив к ферментативному разрушению (в том числе и для аргининаминопептидазы плаценты, т.е. его можно применять при гестагенном типе несахарного диабета), что обусловлено изменениями в структуре молекулы.
В настоящее время десмопрессин выпускается в различных фармацевтических формах. Препарат применяют 2-3 раза в сутки в начальной дозе 0,1 мг для таблеток, 60 мкг для таблеток подъязычных, или 1-2 раза в сутки в начальной дозе 10 мкг (1 доза) для интраназального дозированного спрея и 5-10 мкг (1-2 капли) для интраназальных капель. Затем дозу препарата изменяют до достижения оптимальной - минимальной (для каждой отдельной лекарственной формы десмопрессина) для контроля избыточной жажды и полиурии. для контроля избыточной жажды и полиурии.
Лечение врожденного нефрогенного несахарного диабета проводят с помощью тиазидных диуретиков [гидрохлоротиазид (гипотиазид♠ ) 50-100 мг/сут] и НПВС (индометацин 25-75 мг/сут, ибупрофен 600-800 мг/сут) или комбинацией этих препаратов. При приобретенном нефрогенном несахарном диабете в первую очередь проводят лечение сопутствующего заболевания.
Дальнейшее ведение
В связи с тем, что терапию десмопрессином в основном подбирают по самочувствию пациента, компенсация заболевания зависит от функциональной сохранности центра жажды. При этом рекомендуют периодическое определение осмоляльности плазмы крови и/или концентрации натрия крови, измерение АД, определение наличия отеков для исключения передозировки/недостаточности препарата. Наиболее тяжелые пациенты - больные с нарушениями чувства жажды. Питьевой режим при адипсическом варианте таких нарушений рекомендуют или фиксированный, или зависящий от объема выделенной мочи. При выраженном дипсогенном компоненте несахарного диабета (НЕ при первичной полидипсии!) возможно также прерывистое назначение десмопрессина, т.е. с периодическим пропуском дозы препарата для предупреждения развития водной интоксикации. Отсутствие патологии гипоталамо-гипофизарной области по данным МРТ при центральной форме несахарного диабета не отрицает необходимости повторного проведения исследования в динамике.
Целесообразна оценка МРТ через 1, 3 и 5 лет при условии отсутствия отрицательной динамики со стороны неврологической симптоматики и полей зрения, так как центральный несахарный диабет может предшествовать обнаружению опухолей гипоталамо-гипофизарной области на несколько лет.
СПИСОК ЛИТЕРАТУРЫ
-
Дзеранова Л.К., Пигарова Е.А. Центральный несахарный диабет: современные аспекты диагностики и лечения // Лечащий врач. - 2006. - №10. - С. 44-51.
-
Jane J.A., Vance M.L., Laws E.R. Neurogenic diabetes insipidus // Pituitary. - 2006.
-
Robertson G.L. Diabetes insipidus // Endocrinology Metab. Clin. N. Am. - 1995. - Vol. 24. - P. 549-572.
-
Robinson A.G., Verbalis J.G. The posterior pituitary // Williams textbook of endocrinology; ed. by P.R. Larsen, H.M. Kronenberg, S. Melmed, K.S. Polonsky. - 10th ed. - Philadelphia, 2003. - P. 281-330.
-
Пигарова Е.А. Глава 13. Заболевания нейрогипофиза. Клиническая нейроэндо-кринология / Под ред. И.И. Дедова. - М.: УП Принт, 2011. - С. 239-256.
ПРОЛАКТИНОМА
Романцова Т.И.
ОБЩЕПРИНЯТОЕ НАЗВАНИЕ, СИНОНИМЫ, КОД ПО МКБ-10, ОПРЕДЕЛЕНИЕ ЗАБОЛЕВАНИЯ
Пролактинома, синоним - пролактин-секретирующая аденома гипофиза. Код по МКБ-10: Е22.1. Гиперпролактинемия.
Пролактинома - опухоль (аденома) гипофиза, вырабатывающая пролактин. Пролактиномы - доброкачественные образования (в литературе описаны единичные случаи малигнизации). Основным проявлением пролактином является нарушение функции репродуктивной системы, при больших размерах аденомы - неврологическая симптоматика.
Классификация гиперпролактинемического гипогонадизма - см. «Синдром гиперпролактинемии».
ОСНОВНЫЕ ЧЕРТЫ БОЛЕЗНИ
Пролактиномы, как и подавляющее большинство новообразований гипофиза, имеют моноклональную природу и возникают в результате генетических нарушений в примордиальной (слово «primordialis» в переводе с латинского обозначает «первоначальный») стволовой клетке в период ее дифференцировки. В дальнейшем происходит опухолевая трансформация клетки с последующей клональной экспансией, что приводит к образованию аденомы.
Во время индукции онкогенеза мутация, возникшая спонтанно, или активирует какой-либо протоонкоген, или подавляет ген-супрессор опухолевого роста. Мутации могут подвергнуться не только стволовые клетки, но и зрелые. При этом иногда происходит частичная дедифференцировка лактотрофов (клетки, вырабатывающие пролактин) без значительных иммуногистохимических изменений. В результате возникает клон аденоматозных клеток, обладающих особыми свойствами. Последующий рост и клональная экспансия этих клеток происходят под влиянием гипоталамических гормонов, паракринных факторов роста, а также вследствие нарушения баланса между ними.
В редких случаях пролактиномы могут быть одним из проявлений наследственно обусловленного заболевания - синдрома МЭН-1 либо диагностироваться в рамках семейных изолированных пролактином.
Симптоматика, обусловленная повышением уровня пролактина в сыворотке крови и наличием опухолевого процесса в гипоталамо-гипофизарной зоне, изложена в разделе «Синдром гиперпролактинемии».
ЭПИДЕМИОЛОГИЯ
Пролактиномы составляют наиболее распространенную группу гипофизарных опухолей (порядка 40% всех гормонально-активных новообразований гипофиза). Среди взрослого населения пролактиному диагностируют в 100 случаях из 1 000 000. В первую очередь заболеванию подвержены женщины репродуктивного возраста. В группе пациентов моложе 50 лет количество женщин превышает число мужчин в 10 раз. Однако в пожилом возрасте женщины и мужчины болеют одинаково часто. У детей пролактиномы возникают реже. Они составляют 50% всех опухолей гипофиза, обнаруженных в детском возрасте.
ПРОФИЛАКТИКА
Специфической профилактики гипофизарных опухолей не существует.
КЛАССИФИКАЦИЯ
Классификация пролактином по размеру (рис. 14.5):
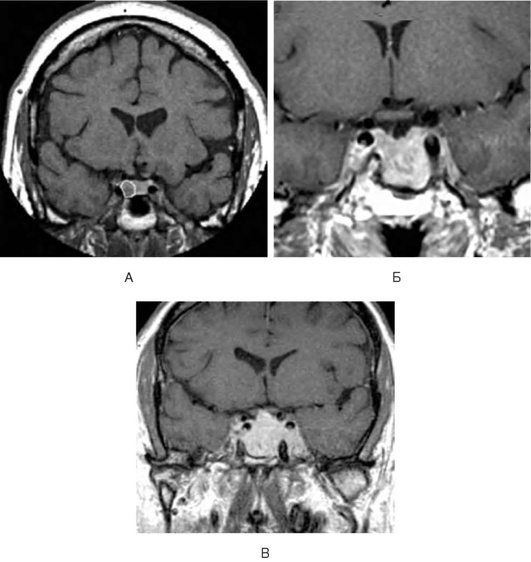
ДИАГНОСТИКА
Необходимые диагностические исследования подробно описаны в разделе «Синдром гиперпролактинемии».
ПРИМЕР ФОРМУЛИРОВКИ КЛИНИЧЕСКОГО ДИАГНОЗА
Гиперпролактинемический гипогонадизм. Макроаденома гипофиза с супраселлярным ростом без нарушения зрительных функций. Вторичная аменорея. Первичное бесплодие.
СКРИНИНГ
Всем пациентам с нарушением функций репродуктивной системы и неоднократно зафиксированным повышением содержания пролактина в сыворотке крови необходимо проведение МРТ головного мозга (при исключении причин вторичной гиперпролактинемии).
ЛЕЧЕНИЕ
Цели лечения
Показания к госпитализации
Медикаментозное лечение
При лечении пролактин-секретирующих опухолей в первую очередь применяют агонисты дофамина, использование которых позволяет нормализовать уровень пролактина, добиться восстановления менструального цикла и фертильной функции у 85-92% женщин репродуктивного возраста. Лечение агонистами дофамина нормализует уровень пролактина и функцию гонад у 80% мужчин. На фоне медикаментозного лечения у 62% больных происходит уменьшение размеров опухоли (рис. 14.6).
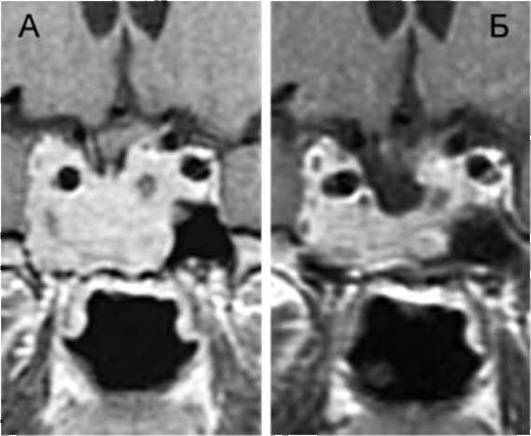
Вероятность и степень опухолевой регрессии зависят от чувствительности организма пациента к препарату, дозы ЛС, вида агониста дофамина, а также от длительности лечения. Применение пролонгированных дофаминомиметиков (каберголин, хинаголид) позволяет получить максимальный лечебный эффект. Положительную динамику иногда наблюдают по истечении нескольких недель после начала медикаментозной терапии. Однако, несмотря на это, пациенты нуждаются в длительном лечении (не менее нескольких лет).
Основные схемы терапии
Для лечения используют агонисты дофамина.
-
Бромокриптин принимают внутрь во время еды по 0,625 мг на ночь; в течение первой недели дозу увеличивают до 2,5 мг. При необходимости производят дальнейшее увеличение дозы на 2,5 мг еженедельно, контролируя при этом уровень пролактина в сыворотке крови. Максимальная доза препарата не должна превышать 12,5 мг в сутки.
-
Хинаголид применяют внутрь: первые 3 дня по 25 мкг 1 раз в сутки перед сном с небольшим количеством пищи, затем каждые 3 дня постепенно повышают дозу на 25 мкг. Через неделю суточная доза составляет 75 мкг. При необходимости производят дальнейшее увеличение дозы на 7,5 мкг ежемесячно, контролируя концентрацию пролактина в сыворотке крови. Максимальная доза препарата не должна превышать 150-300 мкг/сут.
-
Каберголин является наиболее предпочтительным препаратом при лечении пролактином, в том числе в случаях гигантских опухолей (размер от 40 мм). Каберголин принимают внутрь 2 раза в неделю по 0,25-0,5 мг вечером во время еды. При необходимости дальнейшее увеличение дозы производят на 0,5 мг ежемесячно, контролируя концентрацию пролактина в сыворотке крови. Максимальная доза препарата не должна превышать 4 мг в неделю (по 2 мг 2 раза в неделю).
Пролактинома и беременность
Восстановление фертильности и, как следствие, наступление беременности считается одной из важнейших целей лечения пролактин-секретирующих опухолей гипофиза. Если во время применения агонистов дофамина наступила беременность, прием препаратов прекращают. Выраженное повышение содержания эстрогенов может способствовать прогрессии роста опухоли, что проявляется возникновением и усилением головных болей, а также нарушением зрения. Клинически значимая прогрессия роста в период беременности наблюдается у 2,7% больных с микроаденомами, у 22,9% больных с не леченными ранее макроаденомами и у 4,8% больных с макропролактиномами, которым исходно проводилась патогенетическая медикаментозная терапия.
При беременности проводить МРТ головного мозга нежелательно. Поэтому женщинам необходимы консультации невропатолога и офтальмолога в течение всего периода вынашивания плода. Любые вышеуказанные жалобы - повод для внеочередного визита к специалистам. Лучевая терапия и/или хирургическое лечение, выполненные до наступления беременности, полностью не устраняют риск дальнейшего роста опухоли, хотя и несколько снижают его.
Определение концентрации пролактина во время беременности нецелесообразно: у здоровых женщин, так же, как и у пациенток с гиперпролактинемическим гипогонадизмом, содержание гормона достигает крайне высоких значений.
Пролактин влияет на синтез сурфактанта, поэтому превентивное назначение агонистов дофамина в течение всего срока вынашивания плода женщинам с опухолью гипофиза нельзя считать оправданным: нормопролактинемия, достигнутая таким способом, может спровоцировать развитие респираторного дистресс-синдрома у ребенка. Применение дофаминомиметиков обосновано только при подозрении на прогрессирующий рост опухоли (по данным неврологического статуса, клинической картины, результатам периметрии и оценке глазного дна). При отсутствии положительной динамики на фоне консервативного лечения во время беременности пациенткам рекомендовано проведение МРТ головного мозга (без контраста) и срочное решение вопроса о нейрохирургическом вмешательстве. Прогноз беременности можно существенно улучшить применением дофаминергических препаратов в течение года перед планируемым зачатием; во время такой терапии рекомендуется использовать барьерные контрацептивы. В ближайшие 1-2 мес после родов врачи назначают контрольную МРТ. Грудное вскармливание, как правило, не противопоказано. Подавление лактации в послеродовом периоде требуется лишь при явном увеличении опухолевой массы.
Оперативное вмешательство
Показания к хирургическому вмешательству:
В настоящее время при обнаружении дефектов полей зрения проведение операции не требуется, поскольку в большинстве случаев восстановление зрительных функций происходит на фоне медикаментозной терапии.
Наиболее распространенным способом нейрохирургического вмешательства, широко применяемым как при микропролактиномах, так и при макропролактиномах, является транссфеноидальный доступ (через нос). Транскраниально выполняют удаление только больших опухолей с экстраселлярным ростом.
После проведения микроаденомэктомии содержание пролактина нормализуется за 24 часа у 80-90% больных, при этом вероятность рецидива составляет в среднем 17%. После операции по поводу макроаденомы концентрация пролактина снижается у 23-33% пациентов, а частота рецидивов достигает 80%.
Успех операции во многом определяется как опытом хирурга, так и свойствами опухоли. Предикторы успешного оперативного вмешательства: исходный уровень пролактина ниже 200 нг/мл, небольшой размер опухоли (до 10 мм), концентрация пролактина в первые дни после аденомэктомии в пределах 5-10 нг/мл и высокая плотность грануляции клеток опухоли по данным морфофункционального исследования.
Существует вполне определенный риск развития осложнений после транссфеноидальной аденомэктомии: гипопитуитаризм возникает примерно в 25% случаев, несахарный диабет - в 10-18%, а ликворея - в 3-5%. Повреждения внутренней сонной артерии и гипоталамических центров, прободение носовой перегородки, слепота и менингит наблюдаются редко, в среднем в 1-2% случаев. Вероятность летального исхода зависит от того, каким образом производят оперативное вмешательство, и составляет 0,9% для транссфеноидальной и 4-5% для транскраниальной аденомэктомии.
Хирургическое лечение часто сочетают с медикаментозной терапией, которую назначают как до, так и после удаления пролактиномы. Как правило, после операции существенно снижаются объем опухолевой ткани и концентрация пролактина в сыворотке крови, поэтому применение современных дофаминомиметиков может оказаться успешным даже при наличии в анамнезе резистентности к ним.
Лучевая терапия
Лучевую терапию не используют как основной метод лечения пролактином. Лучевую терапию проводят либо в случае отказа больных от оперативного вмешательства, либо при существовании противопоказаний к хирургическому лечению, например при тяжелых сопутствующих соматических патологиях. К основным недостаткам лучевой терапии можно отнести невысокую вероятность достижения нормопролактинемии (40-50%) и длительный (до 20 лет) период, предшествующий развитию желаемого эффекта, в течение которого пациенты вынуждены постоянно принимать агонисты дофамина. В связи с небольшой эффективностью, а также высокой вероятностью развития осложнений (гипопитуитаризм, повреждение черепных нервов, повышенный риск инсультов, образование вторичных опухолей) лучевую терапию следует проводить не всегда даже в случае послеоперационных рецидивов.
Дальнейшее ведение, прогноз
Как правило, медикаментозная терапия назначается на длительный срок (иногда - пожизненно), хотя возможны спонтанные ремиссии заболевания. В период подбора оптимальной дозы дофаминомиметиков уровень пролактина в сыворотке крови определяют ежемесячно, после достижения нормопролактинемии - 1 раз в 6 мес. МРТ головного мозга обычно проводят 1 раз в год.
Учитывая возможность ремиссии, плановая отмена препаратов для контроля над содержанием пролактина возможна 1 раз в 2 года.
СПИСОК ЛИТЕРАТУРЫ
-
Дедов И.И., Мельниченко Г. А, Дзеранова Л.К. и др. Федеральные клинические рекомендации по гиперпролактинемии: клиника, диагностика, дифференциальная диагностика и методы лечения // Пробл. эндокринол. - 2013. - №6. - С. 19-26.
-
Дедов И.И., Мельниченко Г.А., Романцова Т.И. Синдром гиперпролактинемии. - М.: Триада, 2004. - 304 с.
-
Мельниченко Г.А., Марова Е.И., Дзеранова Л.К. и др. Гиперпролактинемия у женщин и мужчин: Пособие для врачей. - М., 2007. - 56 с.
-
Casanueva F.F., Molitch M.E., Schlechte J.A. et al. Gudelines of the Pituitary Society for the diagnosis and management of prolactinomas // Clin. Endocrinol. - 2006. - Vol. 65. - Р. 265-273.
-
Colao A. Pituitary tumours: the prolactinoma // Best Pract. Res. Clin. Endocrinol. Metab. - 2009. - Vol. 23 (5). - P. 575-596.
-
Jillam M.P., Molitch M.E., Lombardi G. et al. Advances in the treatment of prolactinomas // Endocrine Reviews. - 2006. - Vol. 27 (5). - Р. 485-534.
-
Klibanski A. Prolactinomas // N. Engl. J. Med. - 2010. - Vol. 362 (13). - P. 1219- 1226.
-
Kars M., Dekkers O.M., Pereira A.M., Romijn J.N. Update in prolactinomas // Neth. J. Med. - 2010. - Vol. 68 (3). - P. 104-112.
-
Melmed S., Casanueva F.F., Hoffman A.R. et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline // J. Clin. Endocrinol. Metab. - 2011. - Vol. 96 (2). - P. 273-288.
-
Molitch M.E. Mamagement of pregnant patient with a prolactinoma // Eur. J. Endocrinol. - 2015. - Vol. 172. - P. 205-213.