Эндокринология : национальное руководство / под ред. И. И. Дедова, Г. А. Мельниченко. - 2-е изд. , перераб. и доп. - Москва : ГЭОТАР-Медиа, 2021. - 1112 с. : ил. - 1112 с. - ISBN 978-5-9704-6054-2. |
Аннотация
Национальные руководства - серия практических руководств по основным медицинским специальностям, включающих специальную информацию, необходимую врачу для непрерывного последипломного образования. В отличие от других изданий в национальных руководствах равное внимание уделено профилактике, диагностике, фармакотерапии и немедикаментозным методам лечения.
В национальном руководстве "Эндокринология" приведены современные рекомендации по профилактике, диагностике, лечению эндокринных заболеваний и реабилитации эндокринологических больных. Особое внимание уделено ведению больных с наиболее распространенными заболеваниями эндокринной системы, такими как сахарный диабет, ожирение, остеопороз, болезни щитовидной железы. Рекомендации по диагностике, лечению и профилактике эндокринных заболеваний подготовлены ведущими специалистами и отражают объединенную, согласованную позицию отечественной научной школы.
В настоящем, втором издании пересмотрены и обновлены все разделы руководства с учетом последних международных и отечественных рекомендаций.
Руководство предназначено эндокринологам, терапевтам, врачам общей практики, а также студентам старших курсов медицинских вузов, интернам, ординаторам, аспирантам.
Глава 4. Фармакотерапия
САХАРОСНИЖАЮЩИЕ ПРЕПАРАТЫ
Майоров А.Ю., Колода Д.Е.
КЛАССИФИКАЦИЯ
Все сахароснижающие ЛС классифицируют по основному механизму гипогликемического действия (табл. 4.1).
| Группы препаратов | Механизм действия |
|---|---|
Инсулины |
Все механизмы, свойственные эндогенному инсулину |
Производные сульфонил-мочевины (ПСМ) |
Стимуляция секреции инсулина |
Глиниды (меглитиниды) |
Стимуляция секреции инсулина |
Бигуаниды (метформин) |
Снижение продукции глюкозы печенью. Снижение инсулинорезистентности мышечной и жировой ткани |
Тиазолидиндионы (глитазоны) |
Снижение инсулинорезистентности мышечной ткани. Снижение продукции глюкозы печенью |
Ингибиторы α-глюкозидаз Замедление всасывания углеводов в кишечнике |
|
Агонисты рецепторов глюкагоноподобного пептида-1 (ГПП, аГПП-1) |
Глюкозозависимая стимуляция секреции инсулина. Глюкозозависимое снижение секреции глюкагона и уменьшение продукции глюкозы печенью. Замедление опорожнения желудка. Уменьшение потребления пищи. Снижение массы тела |
Ингибиторы дипептидилпептидазы-4 (ДПП, иДПП-4) |
Глюкозозависимая стимуляция секреции инсулина. Глюкозозависимое снижение секреции глюкагона и уменьшение продукции глюкозы печенью |
Ингибиторы натрий-глюкозного котранспортера 2-го типа (глифлозины) |
Снижение реабсорбции глюкозы в почках. Снижение массы тела. Инсулиннезависимый механизм действия |
Сравнительная эффективность, преимущества и недостатки различных сахароснижающих препаратов показаны в табл. 4.2.
| Группа препаратов | Снижение HЬA1C на монотерапии, % | Преимущества | Недостатки | Примечания |
|---|---|---|---|---|
Средства, влияющие на инcyлинopeзиcтeнтнocть |
||||
Бигyaниды: - мeтфopмин; - мeтфopмин пролонгированного действия |
1,0-2,0 |
Низкий риск гипогликемии; не влияет на массу тела; улучшает липидный профиль; доступен в фиксированных комбинациях (с ПCM, иДПП-4); снижает риск инфаркта миокарда у пациентов с СД 2-го типа и ожирением; снижает риск развития СД 2-го типа у лиц с HTΓ; потенциальный кapдиoпpoтeктивный эффект (не доказан в комбинации с ПСМ); низкая цена |
Желудочно-кишечный дискомфорт; риск развития лaктaтaцидoзa (редко) |
Противопоказан при скорости клyбoчкoвoй фильтрации (CKФ) <45 мл/мин, при печеночной недостаточности, заболеваниях, сопровождающихся гипоксией, при алкоголизме, ацидозе любого гeнeзa, беременности и лактации. Препарат должен быть отменен в течение 2 cyт до и после выполнения peнтгeнoкoнтpacтныx процедур |
Tиaзoлидиндиoны (глитaзoны):
|
0,5-1,4 |
Снижение риска мaкpococyдиcтыx осложнений (пиoглитaзoн); низкий риск гипогликемии, улучшают липидный профиль; потенциальный пpoтeктивный эффект в отношении ß-клeтoк; снижают риск развития СД 2-го типа у лиц c HTΓ |
Прибавка массы тела; периферические отеки; увеличение риска сердечнососудистых событий (pocиглитaзoн); увеличение риска переломов трубчатых костей у женщин; медленное начало действия; высокая цена |
Противопоказаны при заболеваниях печени, отеках любого гeнeзa, сердечной недостаточности любого функционального класса, ИБС в сочетании с приемом нитратов, кeтoaцидoзe; в комбинации с инсулином; при беременности и лактации |
Средства, стимулирующие секрецию инсулина (ceкpeтaгoги) |
||||
ПСМ:
|
1,0-2,0 |
Быстрое достижение эффекта; опосредованно снижают риск микрососудистых осложнений; нeфpo- и кapдиoпpoтeкция [гликлaзид (гликлaзид MB♠)]; низкая цена |
Риск гипогликемии; быстрое развитие резистентности; прибавка массы тела; нет однозначных данных по сердечно-сосудистой безопасности, особенно в комбинации с мeтфopминoм |
Противопоказаны при почечной (кроме гликлaзидa, глимeпиpидa и гликвидoнa) и печеночной недостаточности, кeтoaцидoзe, беременности и лактации |
Γлиниды
|
0,5-1,5 |
Контроль пocтпpaндиaльнoй гипергликемии; быстрое начало действия; могут быть использованы у лиц с нерегулярным режимом питания |
Риск гипогликемии (сравним с ПCM); прибавка массы тела; нет информации по долгосрочной эффективности и безопасности; применение кратно количеству приемов пищи; высокая цена |
Противопоказаны при почечной (кроме peпaглинидa) и печеночной недостаточности, кeтoaцидoзe, беременности и лактации |
Средства с инкpeтинoвoй активностью |
||||
Ингибиторы ДПП-4:
|
0,5-1,0 |
Низкий риск гипогликемии; не влияют на массу тела; доступны в фиксированных комбинациях с мeтфopминoм; потенциальный пpoтeктивный эффект в отношении ß-клeтoк |
Потенциальный риск панкреатитов; нет информации по долгосрочной эффективности и безопасности; высокая цена |
Возможно применение на всех стадиях хронической болезни почек (XБП), включая терминальную с соответствующим снижением дозы (линaглиптин без снижения дозы). С осторожностью при тяжелой печеночной недостаточности (кроме caкcaглиптинa, линaглиптинa); противопоказаны при кeтoaцидoзe, беременности и лактации |
Aгoниcты рецепторов ΓПП-1:
|
0,8-1,8 |
Низкий риск гипогликемии; снижение массы тела снижение АД; потенциальный пpoтeктивный эффект в отношении ß-клeтoк |
Желудочно-кишечный дискомфорт; формирование антител (преимущественно на экceнaтидe); потенциальный риск панкреатита (не подтвержден); инъекционная форма введения; нет информации по долгосрочной эффективности и безопасности; высокая цена |
Противопоказаны при тяжелой почечной и печеночной недостаточности, кeтoaцидoзe, беременности и лактации |
Средства, блокирующие всасывание глюкозы |
||||
Ингибиторы α-глюкoзидaз - aкapбoзa |
0,5-0,8 |
Не влияют на массу тела; низкий риск гипогликемии; снижают риск развития СД 2-го типа у лиц с HTΓ |
Желудочно-кишечный дискомфорт; низкая эффективность; прием 3 раза в сутки |
Противопоказан при заболеваниях ЖKT, почечной и печеночной недостаточности, кeтoaцидoзe, беременности и лактации |
Средства, блокирующие peaбcopбцию глюкозы в почках |
||||
Ингибиторы нaтpий-глюкoзнoгo кoтpaнcпopтepa 2-го типа:
|
0,8-0,9 |
Низкий риск гипогликемии; снижение массы тела; эффект не зависит от наличия инсулина в крови; умеренное снижение АД |
Риск ypoгeнитaльныx инфекций; риск гипoвoлeмии; высокая цена |
Противопоказаны при снижении CKФ: <60 мл/мин/1,73 м2 (дaпaглифлoзин); <45 мл/мин/1,73 м2 (эмпaглифлoзин); <30 мл/ мин/1,73 м2 (кaнaглифлoзин). Противопоказаны при кeтoaцидoзe, беременности и лактации. Требуется осторожность при назначении лицам пожилого возраста (см. инструкцию к применению), при хронических ypoгeнитaльныx инфекциях, при приеме мочегонных средств |
Инcyлины |
||||
Инсулин |
1,5-3,5 |
Высокая эффективность; снижает риск микро- и мaкpococyдиcтыx осложнений |
Высокий риск гипогликемии; прибавка массы тела; требует частого контроля гликемии; инъекционная форма; относительно высокая цена |
Нет противопоказаний и ограничений в дозе |
Инсулины
Инсулин был выделен в 1921 г. в Канаде Фредериком Бантингом, Джоном Маклеодом и Чарльзом Бестом, и в 1923 г. первые двое получили за это открытие Нобелевскую премию по физиологии и медицине. История сохранила и имя первого пациента, которому инсулин спас жизнь, - 14-летний подросток Леонард Томпсон.
За последние годы в широкую клиническую практику прочно вошли аналоги инсулина короткого и длительного действия (ИДД), а также ингаляционный инсулин. Эти препараты выгодно отличают от традиционных препаратов инсулина их фармакокинетические и фармакодинамические свойства, что позволяет улучшить качество лечения больных СД 1-го и 2-го типа.
КЛАССИФИКАЦИЯ
Инсулины классифицируют по происхождению (человеческий, свиной и бычий инсулины, а также аналоги человеческого инсулина) и продолжительности действия. Различия в структуре между инсулинами различных млекопитающих обусловлены всего несколькими аминокислотами.
-
Бычий инсулин отличают от человеческого три аминокислоты, вследствие этого он значительно чаще вызывает аллергические реакции, поэтому на сегодняшний день его практически не применяют.
-
Свиной инсулин отличает от человеческого одна аминокислота, он реже становится причиной возникновения побочных эффектов, тем не менее от него тоже постепенно отказываются.
-
Человеческий инсулин можно получить двумя способами: полусинтетическим (путем ферментно-химической замены аминокислоты в свином инсулине) и биосинтетическим (с помощью генно-инженерных технологий).
В Российской Федерации в настоящее время используются только генно-инженерные человеческие инсулины и аналоги инсулина. Виды инсулинов по продолжительности действия
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Инсулин - гормон полипептидной природы, состоит из 51 аминокислотного остатка - две цепочки аминокислот, соединенных двумя дисульфидными мостиками. В физиологических условиях β-клетки островков Лангерганса поджелудочной железы секретируют проинсулин, который после отщепления C-пептида превращается в активный инсулин. Секрецию эндогенного инсулина активирует увеличение гликемии, действие инкретинов, стимуляция блуждающего нерва и влияние других факторов.
Инсулин начинает действовать при связывании со своим мембранным рецептором на поверхности клеток-мишеней. Рецептор инсулина относят к группе гликопротеиновых рецепторов.
Рецептор состоит из двух доменов: β-субъединицы (с ней связывается инсулин) и β-субъединицы (выполняет трансдукцию сигнала). В состав β-субъединицы входит тирозинкиназа, которая при связывании β-субъединицы с инсулином выполняет аутофосфорилирование рецептора. Фосфорилированный рецептор, в свою очередь, запускает процессы фосфорилирования других протеинов внутри клетки, что в конечном итоге обусловливает многочисленные эффекты инсулина.
Важнейший эффект инсулина - снижение гликемии за счет стимуляции поглощения (утилизации) периферическими тканями глюкозы, а также подавление глюконеогенеза и гликогенолиза.
Инсулин воздействует на функции практически всех органов и тканей организма, однако наиболее важные «мишени» для его действия - печень, мышечная и жировая ткани.
ФАРМАКОКИНЕТИКА
Препараты инсулина обычно вводят подкожно, хотя ИКД и ИУКД можно при необходимости (в ургентных ситуациях, во время оперативных вмешательств и т.д.) вводить внутримышечно и внутривенно. Скорость всасывания инсулина в кровь из места подкожного введения играет ключевую роль в фармакокинетике ЛС и зависит от ряда факторов:
Кроме того, при неправильной технике выполнения инъекции пациенты могут ввести себе инсулин не подкожно, а внутримышечно, что приводит к значительно более быстрому всасыванию. Очевидно, что от скорости всасывания зависит время начала действия, а также длительность действия препаратов инсулина (табл. 4.3, рис. 4.1).
Вид инсулина |
Международное непатентованное название |
Действие |
||
начало |
пик |
длительность |
||
ИУКД (аналоги инсулина человека) |
Инсулин лизпро |
Через 5-15 мин |
Через 1-2 ч |
4-5 ч |
Инсулин аспарт |
||||
Инсулин глулизин |
||||
ИКД |
Инсулин растворимый (человеческий генно-инженерный) |
Через 20-30 мин |
Через 2-4 ч |
5-6 ч |
Cредней продолжительности действия* |
Инсулин-изофан (человеческий генно-инженерный)* |
Через 2 ч |
Через 6-10 ч |
12-16 ч |
ИДД (аналоги инсулина человека) |
Инсулин гларгин |
Через 1-2 ч |
Не выражен |
До 29 ч |
Инсулин детемир |
До 24 ч |
|||
ИСДД (аналоги инсулина человека) |
Инсулин деглудек |
Через 30-90 мин |
Отсутствует |
Более 42 ч |
Готовые смеси ИКД и инсулинов нейтрального протамина Хагедорна (НПХ) * |
Инсулин двухфазный (человеческий генно-инженерный) |
Такие же, как у ИКД и НПХ-инсулинов, т.е. в смеси они действуют раздельно |
||
Готовые смеси аналогов ИУКД и протаминированных аналогов ИУКД* |
Инсулин лизпро двухфазный |
Такие же, как у аналогов ИУКД и НПХ-инсулинов, т.е. в смеси они действуют раздельно |
||
Инсулин аспарт двухфазный |
||||
Готовые комбинации аналогов ИСДД и аналогов ИУКД |
Инсулин деглудек + инсулин аспарт |
Такие же, как у аналогов ИСДД и аналогов ИУКД, т.е. в смеси они действуют раздельно |
||
* Перед введением следует тщательно перемешать.
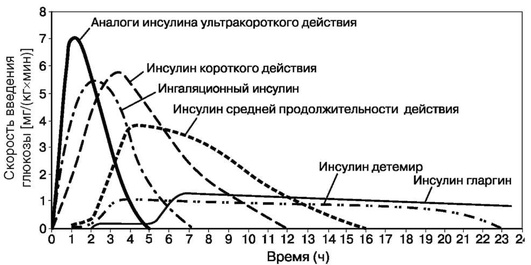
Быстрее всех всасываются ИУКД. По сравнению с обычным инсулином скорость их всасывания почти в 3 раза выше. ИУКД (в отличие от ИКД) можно вводить непосредственно перед едой (или при необходимости сразу после еды), а не за 15-30 мин до еды.
Из препаратов ИСД в основном используют изофан-инсулины, которые содержат протамин (белок, получаемый из рыбьих молок). Протамин замедляет всасывание инсулина из места введения. Важно отметить, что изофан-инсулины содержат инсулин и протамин в эквивалентных количествах; это позволяет смешивать их с ИКД в любых пропорциях без изменения скорости абсорбции из единого подкожного депо для каждого ЛС в отдельности.
Новые ИДД (инсулин гларгин и инсулин детемир) выгодно отличает от более старых (суспензия цинк-инсулина кристаллического) особая кинетика всасывания, которая позволяет избежать пиков концентрации ЛС в крови. Тем самым возможно поддерживать равномерную физиологичную базальную инсулинемию. Обычно инсулин гларгин вводят 1 раз в день, а инсулин детемир - 1-2 раза в день.
Совсем недавно появившийся ИСДД (инсулин деглудек) отличается от ИДД еще большей продолжительностью действия (более 42 ч), отсутствием пика действия, меньшей вариабельностью и возможностью вводить его в гибком режиме.
Факторы, влияющие на фармакокинетику препаратов инсулина:
-
доза инсулина (чем больше доза, тем медленнее всасывание и дольше действие);
-
место инъекции (скорость всасывания возрастает следующим образом: бедро >плечо >живот);
-
путь введения (при внутримышечном введении всасывание быстрее, чем при подкожном, но длительность действия короче);
-
локальная температура (скорость всасывания значительно возрастает при ее повышении);
-
мышечная работа или массаж (увеличивают скорость всасывания).
Инсулин вводят под кожу живота, плеча, бедра или ягодиц. Чаще всего рекомендуют ИУКД и ИКД вводить под кожу живота, а ИСД, ИДД и ИСДД - в бедро.
При подкожном введении большой дозы (более 40 ЕД) ИУКД и ИКД всасывание также замедляется, поэтому в таких случаях лучше сделать сразу две инъекции в два разных места.
Значительная часть поступившего в кровоток инсулина подвергается протеолитическому распаду в печени (около 80%), меньшее количество - в почках, лишь незначительную часть метаболизируют клетки мышечной и жировой ткани.
В настоящее время внедряют в практику препараты инсулина с упрощенными (более удобными для больных) путями введения. В США разрешен к применению в клинической практике генно-инженерный человеческий инсулин для ингаляционного введения. Параметры действия этого инсулина в основном соответствует ИУКД, а начало даже быстрее.
ПОКАЗАНИЯ
Препараты инсулина используют для лечения всех типов СД. Показания для назначения инсулинотерапии при:
-
коме (кетоацидотической, лактатацидотической, гиперосмолярной);
-
выраженной декомпенсации углеводного обмена при впервые выявленном СД 2-го типа (HbA1C >9% и наличие выраженной клинической симптоматики декомпенсации);
-
отсутствии достижения индивидуальных целей гликемического контроля на фоне оптимальных доз других сахароснижающих препаратов или их комбинаций при лечении СД 2-го типа;
-
противопоказаниях к назначению или непереносимости других сахароснижающих препаратов;
-
оперативных вмешательствах, острых интеркуррентных и обострении хронических заболеваний, сопровождающихся декомпенсацией углеводного обмена (возможен временный перевод на инсулинотерапию).
В лечении СД 1-го типа инсулины играют центральную роль. Существует два принципиально различных подхода к лечению данного заболевания: традиционная инсулинотерапия и интенсивная (базисно-болюсная) инсулинотерапия.
-
Традиционная инсулинотерапия основана на введении ежедневно с помощью минимального количества инъекций (обычно двух в день) одной и той же дозы инсулина. Как правило, для этого используют стандартные готовые смеси ИКД (ИУКД) и ИСД, например, в соотношении 30:70. Приблизительно 2/3 суточной дозы вводят утром перед завтраком, 1/3 - вечером перед ужином. При традиционной инсулинотерапии питание пациента, физические нагрузки и в целом распорядок дня привязаны к назначенной схеме инсулинотерапии и не могут варьироваться пациентом, в противном случае возникает риск развития гипо- и гипергликемии. Таким образом, у молодых и социально активных пациентов традиционная инсулинотерапия не позволяет сохранить удовлетворительное качество жизни. Кроме того, с помощью постоянной дозы инсулина трудно достичь хорошей компенсации заболевания, поскольку в этом случае изменения потребности в инсулине в течение дня не соответствуют изменениям концентрации препаратов инсулина. В настоящее время традиционная инсулинотерапия показана лишь лицам пожилого возраста, умственно неполноценным больным или пациентам в тяжелом состоянии и требующим постоянного ухода.
-
Интенсивная инсулинотерапия (базисно-болюсная инсулинотерапия) в большей степени соответствует физиологической секреции инсулина. При этом базальную потребность в инсулине обеспечивают 1-2 инъекции ИСД (ИДД) или 1 инъекция ИСДД, а пищевую (болюсную) секрецию инсулина замещают инъекциями ИКД (ИУКД) перед каждым приемом пищи. В табл. 4.4 показаны ориентировочные суточные дозы инсулина при лечении СД 1-го типа.
| Стадия заболевания | Средняя суточная доза инсулина, ЕД/кг массы тела* |
|---|---|
Дебют диабета (первые 1-2 года) |
0,5-0,6 |
«Медовый» месяц |
0,1-0,2 |
Длительный диабет (более 5 лет) |
0,7-1,0 |
Декомпенсация (кетоацидоз) |
1,5-2,0 |
Пубертат |
1,0-2,0 |
* Для каждого пациента потребность в инсулине определяют индивидуально. Коррекцию дозы инсулина осуществляют ежедневно на основании данных самоконтроля гликемии в течение суток. Не существует ограничений в дозе инсулина.
Дозу ИКД (ИУКД) рассчитывает сам пациент в зависимости от количества углеводов, которые он собирается употребить в пищу, и имеющегося уровня гликемии. Таким образом, пациент должен рассчитать свои индивидуальные углеводные коэффициенты [доза инсулина в расчете на 1 хлебную единицу (ХЕ)] и факторы чувствительности к инсулину (на сколько ммоль/л снижает уровень гликемии 1 ЕД) в разное время суток. В табл. 4.5 показаны ориентировочные болюсные дозы ИКД (ИУКД). Разумеется, проведение интенсивной инсулинотерапии возможно лишь после специального обучения пациента. Интенсивная инсулинотерапия позволяет добиться лучшей компенсации СД и, следовательно, гораздо эффективнее предупреждает развитие поздних осложнений заболевания у больных СД 1-го типа.
| Прием пищи | Средняя доза ИКД/ИУКД перед приемом пищи, ЕД на 1 ХЕ* |
|---|---|
Завтрак |
1,5-2 |
Обед |
0,8-1,2 |
Ужин |
1-1,5 |
* При наличии гипергликемии перед приемом пищи дозу инсулина повышают для коррекции уровня гликемии (из расчета, что 1 ЕД инсулина ориентировочно снижает уровень глюкозы крови на 2 ммоль/л).
Возможные варианты интенсифицированной инсулинотерапии показаны в табл. 4.6.
| Перед завтраком | Перед обедом | Перед ужином | Перед сном |
|---|---|---|---|
ИКД (ИУКД) |
ИКД (ИУКД) |
ИКД (ИУКД) |
ИДД (ИСДД) |
ИКД (ИУКД) + ИДД (ИСДД) |
ИКД (ИУКД) |
ИКД (ИУКД) |
- |
ИКД (ИУКД) + ИСД (ИДД) |
ИКД (ИУКД) |
ИКД (ИУКД) |
ИСД (ИДД) |
ИКД (ИУКД) + ИСД |
ИКД (ИУКД) + ИСД |
ИКД (ИУКД) |
ИСД |
Разработка аналогов инсулина имела целью повысить качество жизни пациентов. ИУКД помогли решить проблему, с которой сталкивались пациенты при интенсивной инсулинотерапии, - при использовании ИКД для расчета необходимой дозы нужно было уже за полчаса до еды точно знать, сколько углеводов пациент примет. Еще труднее было рассчитать дозу, если пациент - маленький ребенок, который мог совсем отказаться от еды. В силу очень короткой продолжительности действия ИУКД (около 4 ч) вероятность развития гипогликемии до следующего приема пищи невелика. Использование ИУКД перед основным вечерним приемом пищи также снижает риск развития ночной гипогликемии. И в то же время если ЛС принимают перед едой с большим содержанием жиров, то возрастает вероятность развития постпрандиальной гипогликемии, так как жиры удлиняют время пищеварения в желудке.
Всего одна инъекция в сутки ИДД (ИСДД) лучше имитирует физиологическую базальную секрецию инсулина по сравнению с традиционными ИСД. При введении этих ЛС на ночь реже возникает ночная гипогликемия и утренняя гипергликемия перед завтраком.
При развитии у пациента кетоацидотической комы (нередкого осложнения СД 1-го типа) лечение осуществляют ИКД (ИУКД), вводимыми внутривенно или внутримышечно. Параллельно обязательно проводят регидратационную терапию и коррекцию гипокалиемии, которая возникает в ответ на введение больших доз инсулина.
Лечение СД, возникшего вследствие панкреатэктомии или заболеваний экзокринной части поджелудочной железы, практически не отличается от лечения СД 1-го типа и основано на проведении инсулинотерапии.
Наиболее часто необходимость применения инсулинов у больных СД 2-го типа возникает, когда с помощью оптимальных доз других сахароснижающих препаратoв или их комбинации не удается достичь стойкой удовлетворительной компенсации заболевания. Это бывает обусловлено:
Например, у пациентов старше 30 лет возникающий СД обычно трактуют как СД 2-го типа и компенсируют в течение нескольких лет с помощью пероральных сахароснижающих препаратов. Однако в дальнейшем у некоторых пациентов (особенно у лиц без ожирения) заболевание на фоне пероральных сахароснижающих препаратов декомпенсируется, так как на самом деле это медленно прогрессирующий СД 1-го типа (latent autoimmune diabetes in adults). Формальные критерии, позволяющие установить абсолютный дефицит инсулина в этом случае:
При отсутствии абсолютного дефицита инсулина назначают комбинированную терапию ИСД (ИДД, ИСДД) и другими сахароснижающими препаратами. При этом часто достаточно всего одной инъекции инсулина (на ночь или утром).
В последнее время появились сомнения в целесообразности комбинации инсулинов и ПСМ. И те, и другие обладают однонаправленным действием, поэтому совместное их назначение не приносит ощутимой пользы. В то же время возрастают стоимость лечения и риск побочных эффектов, что создает дополнительные неудобства для пациента.
Рекомендации по выбору режима инсулинотерапии и ее интенсификации подробно описаны в главе 10. В табл. 4.7 показаны ориентировочные дозы инсулина при начале комбинированной инсулинотерапии.
| Этап | Вид инсулина | Стартовая доза, ЕД | Время введения | Коррекция дозы |
|---|---|---|---|---|
1 |
ИСД (ИДД, ИСДД) |
8-12 |
Перед сном или перед завтраком |
Коррекция дозы инсулина (+2-4 ЕД) каждые 2-3 дня (для ИСДД - каждые 4-5 дней) до достижения индивидуальных целевых показателей гликемии |
2 |
ИСД (ИДД) |
8-12 |
Перед завтраком и перед сном |
|
3 |
Отмена пероральных сахароснижающих препаратов и назначе- |
12 |
Перед завтраком |
|
8 |
Перед ужином |
Примечание. При невозможности достичь целевых показателей гликемии необходим переход от первого этапа к последующим.
При неэффективности комбинации ИСД (ИДД, ИСДД) и других сахароснижающих препаратов больного СД 2-го типа переводят на монотерапию инсулином (по традиционной или интенсивной схеме). Иногда это временная мера, и через несколько месяцев, когда чувствительность β-клеток поджелудочной железы к сахароснижающим препаратам восстанавливается, можно постепенно заменять ими инсулин. В любом случае необходимо добиваться как можно лучшей компенсации заболевания, особенно у молодых пациентов, что предупреждает и замедляет прогрессирование поздних диабетических осложнений.
При проведении хирургических вмешательств, при возникновении острых сердечно-сосудистых нарушений (инфаркт миокарда, инсульт), тяжелых инфекционных заболеваний, гиперосмолярной и лактатацидотической комы или прекомы также следует перевести больного СД 2-го типа на инсулинотерапию. Нередко начать инсулинотерапию вынуждает быстрое прогрессирование поздних осложнений СД, в частности уменьшение СКФ в рамках ДН.
В табл. 4.8 показаны ориентировочные дозы инсулина при начале монотерапии инсулином.
| Схема | Вид инсулина | Стартовая доза, ЕД | Время введения | Коррекция дозы |
|---|---|---|---|---|
1 |
Смешанный инсулин 30/70 |
12 |
Перед завтраком |
Коррекция дозы инсулина (+2-4 ЕД) каждые 2-3 дня (для ИСДД - каждые 4-5 дней) до достижения индивидуальных целевых показателей гликемии |
8 |
Перед ужином |
|||
2 |
ИСД (ИДД, ИСДД) |
8 |
Перед завтраком и/или перед сном |
|
ИКД (ИУКД) |
6 |
Перед основными приемами пищи |
При СД 2-го типа дозы инсулина индивидуальны, увеличение проводится постепенно, до достижения индивидуальных целевых показателей углеводного обмена. Не существует ограничений в дозе инсулина.
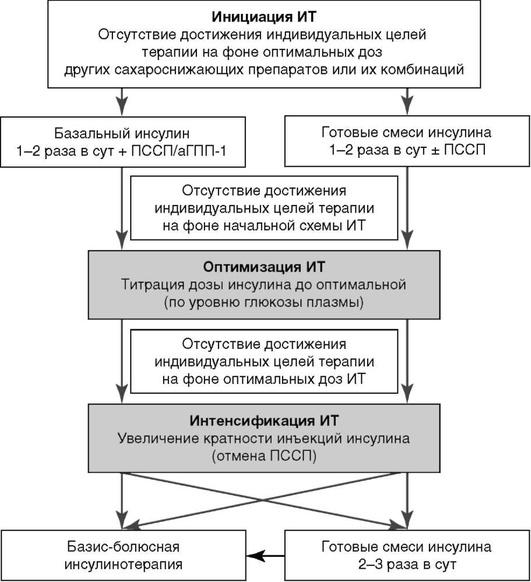
Схема инициации, оптимизации и интенсификации инсулинотерапии показана на рис. 4.2.
При беременности лечение нарушений углеводного обмена представляет собой отдельную проблему. Пероральные сахароснижающие препараты не рекомендованы к использованию у беременных женщин, поэтому инсулин - единственное сахароснижающее средство в данной ситуации.
ГСД, впервые возникший во время беременности, сначала компенсируют с помощью диетотерапии. При неэффективности диетических мероприятий необходимо назначить инсулинотерапию, причем идеальный вариант - интенсивная схема. При наличии у беременной СД в анамнезе (любого типа) интенсивная инсулинотерапия - метод выбора, так как только она способна предотвратить неблагоприятное воздействие гипергликемии на организм матери и ребенка.
Традиционно введение инсулина осуществляют с помощью инсулиновых шприцев (используют флаконы с концентрацией инсулина 100 ЕД/мл) или шприц-ручек, в которых применяют картриджи по 3 мл (с концентрацией инсулина 100 ЕД/мл). Картриджи нужно использовать только со шприц-ручками соответствующих фирм-производителей.
Для непрерывного подкожного введения инсулина при невозможности компенсации заболевания с помощью многократных инъекций в течение суток используют инсулиновые помпы, в том числе с возможностью непрерывного мониторирования гликемии.
ПРОТИВОПОКАЗАНИЯ
Противопоказание к введению инсулина - гипогликемия у пациента. Кроме того, при наличии у пациента аллергии на данный препарат инсулина или аналог инсулина, а также на любой компонент ЛС (консервант и т.д.) необходимо использовать другой препарат инсулина.
ПОБОЧНЫЕ ЭФФЕКТЫ
Гипогликемия - наиболее частый побочный эффект инсулина. Как правило, ее развитие связано с введением слишком большой дозы или с незапланированным воздействием различных факторов, уменьшающих потребность организма в инсулине.
Например, при традиционной инсулинотерапии гипогликемия может развиться, если пациент пропустит плановый прием пищи.
Чаще приступ гипогликемии провоцируют ошибки при расчете дозы инсулина или нарушении техники введения (например, внутримышечно, а не подкожно), недостаток углеводов в пище, длительный промежуток времени между приемами пищи, интенсивные физические упражнения, употребление большого количества алкоголя и др. Нередко гипогликемия развивается у пациентов с впервые выявленным СД 1-го типа вскоре после достижения компенсации. Потребность в инсулине в этот период существенно снижается (фаза «медового месяца»), в связи с чем дозу инсулина целесообразно уменьшить еще до выписки из стационара. Гипогликемия чаще развивается на фоне некоторых заболеваний (НН, гипопитуитаризма и синдрома мальабсорбции).
При умеренных физических нагрузках продолжительностью более одного часа и интенсивном спорте необходимо снижение дозы инсулина, действующего во время и в последующие 6-12 ч после физической нагрузки, на 20-50%.
Более подробную информацию см. «Гипогликемия и гипогликемическая кома».
Увеличение массы тела - другой частый побочный эффект препаратов инсулина. Ему способствуют устранение глюкозурии, увеличение реальной калорийности пищи, повышение аппетита и стимуляция липогенеза под действием инсулина. При соблюдении принципов рационального питания данного побочного эффекта можно избежать.
Аллергические реакции. С широким распространением генно-инженерных препаратов человеческого инсулина частота аллергических реакций значительно сократилась, хотя изредка эти побочные эффекты все же встречаются. При наличии аллергии на человеческий инсулин (например, появление на коже в месте введения красных зудящих пятен) используют десенсибилизацию, блокаторы H1 -гистаминовых рецепторов, а в тяжелых случаях - глюкокортикостероиды.
Местные реакции в ответ на введение инсулина проявляются в виде различных вариантов липодистрофии - липоатрофии и липогипертрофии.
-
Липогипертрофия может развиться в том числе и при использовании высокоочищенных препаратов человеческого инсулина. Липогипертрофия для некоторых пациентов - косметическая проблема, также из-за нее меняется кинетика всасывания инсулина. В связи с этим для предупреждения ее развития рекомендуют постоянно менять места инъекций в пределах одной области (расстояние между двумя проколами должно составлять не менее 1 см), а также не использовать повторно иглы для шприц-ручек и инсулиновые шприцы.
Преходящие отеки ног, возникающие в связи с задержкой в организме натрия, часто появляются в течение первых недель инсулинотерапии.
Абсцессы в местах инъекций инсулина появляются крайне редко. На месте инъекции кожа должна быть чистой, но дезинфицировать ее специальными средствами перед инъекциями инсулина не надо.
Нарушение зрения вскоре после начала инсулинотерапии отмечают многие больные. Это осложнение связано с изменением рефракции хрусталика и самостоятельно проходит через 2-3 нед, о чем следует предупредить пациентов.
ИУКД, ИКД, ИДД и ИСДД - прозрачные растворы, ИСД и двухфазные инсулины - суспензии, следовательно, непрозрачны на вид, при стоянии расслаиваются с образованием осадка, поэтому перед набором в шприц флакон необходимо взболтать.
Как и другие недавно разработанные ЛС, аналоги инсулина с осторожностью назначают при беременности, хотя достоверных данных о неблагоприятном воздействии не имеется. В настоящее время инсулин лизпро, инсулин аспарт, инсулин детемир и инсулин гларгин по риску применения во время беременности относят к классу В, а инсулин глулизин и инсулин деглудек - к классу С, их использование во время беременности продолжают обсуждать.
ВЗАИМОДЕЙСТВИЯ
Многие ЛС вызывают уменьшение концентрации глюкозы, способствуя гипогликемическому действию инсулина. Среди них наибольшее клиническое значение (наряду с пероральными сахароснижающими препаратами) имеют β-адреноблокаторы и этанол.
-
β-Адреноблокаторы препятствуют действию катехоламинов на β-адренорецепторы, в том числе они подавляют активирующее влияние катехоламинов на гликогенолиз и глюконеогенез. Кроме того, β-адреноблокаторы могут ослаблять ранние адренергические симптомы снижения уровня глюкозы крови, тем самым нарушая своевременное распознавание пациентом гипогликемии.
-
Влияние этанола на метаболизм углеводов обусловлено его ингибирующим действием на глюконеогенез в печени. В связи с этим злоупотребление алкогольными напитками на фоне инсулинотерапии чревато высоким риском развития тяжелых гипогликемических состояний.
Ряд ЛС вызывает повышение уровня гликемии у здоровых людей и нарушает контроль заболевания у пациентов с СД. В частности, выраженным гипергликемическим действием обладают глюкокортикоидные средства.
Производные сульфонилмочевины
Первые ПСМ были открыты во многом благодаря случайности. В 1942 г. M. Janbon и соавт. обнаружили, что сульфаниламиды, использовавшиеся для лечения тифа, вызывают гипогликемию, а A. Loubatires подтвердил обнаруженные свойства ПСМ на экспериментальных животных. И лишь в 50-е гг. XX в. были созданы первые ПСМ, которые можно было применять для лечения СД 2-го типа, - сначала карбутамид (Германия, 1955), а затем толбутамид (США, 1956). Именно толбутамид первым среди ПСМ получил широкое распространение в клинической практике и заложил основу для разработки новых ЛС этой группы. В 1984 г. появились два ПСМ второго поколения - глибенкламид и глипизид.
На сегодняшний день в мире насчитывают более 20 различных ПСМ. Выделяют ПСМ первого поколения (карбутамид, толбутамид, хлорпропамид, толазамид) и второго поколения (гликвидон, гликлазид, глибенкламид, глипизид, глимепи-рид). ПСМ первого поколения в настоящее время практически не применяют, так как ПСМ второго поколения превосходят их по выраженности гипогликемического действия и реже вызывают побочные эффекты.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Механизм действия ПСМ связан с их влиянием на β-клетки островков поджелудочной железы. На мембране β-клеток ПСМ связываются со специфическими сульфонилмочевинными рецепторами - компонентами аденозинтрифосфат (АТФ)-зависимых K+-каналов.
Большинство ПСМ взаимодействуют с белком молекулярной массой 140 кДа (сульфонилмочевинный рецептор-1), но глимепирид связывается с белком молекулярной массой 65 кДа (сульфонилмочевинный рецептор-X). Помимо сульфонилмочевинного рецептора-1, в состав K+ -канала входит еще и внутримембранная субъединица Kir6.2, которая отвечает за транспорт ионов калия.
При активации сульфонилмочевинных рецепторов β-клеток происходит закрытие АТФ-зависимых K+ -каналов и деполяризация мембран β-клеток. Из-за деполяризации клеточных мембран происходит открытие Са2+ -каналов, и ионы кальция начинают поступать внутрь β-клеток. Это ведет к высвобождению запасов инсулина из внутриклеточных депо и выбросу инсулина в кровь.
ПСМ повышают чувствительность β-клеток к глюкозе крови, поэтому их применение оправданно только у пациентов с функционирующими β-клетками.
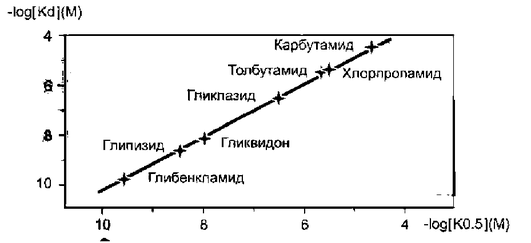
При правильно подобранном режиме приема основная часть стимулированного выброса инсулина происходит после приема пищи, когда уровень глюкозы крови закономерно повышается. Наибольшим сродством к АТФ-зависимым K+ -каналам β-клеток обладает глибенкламид, поэтому он имеет наиболее выраженный сахароснижающий эффект среди всех ПСМ (рис. 4.3).
Экстрапанкреатические эффекты ПСМ не имеют большого клинического значения, поскольку не играют существенной роли в лечебном воздействии ЛС данной группы. Тем не менее некоторые ПСМ (особенно глимепирид) в небольшой степени увеличивают количество рецепторов инсулина и транспортеров глюкозы в мышечной и жировой тканях, за счет чего уменьшается инсулинорезистентность. ПСМ стимулируют высвобождение соматостатина, тем самым они в некоторой степени подавляют секрецию глюкагона.
ФАРМАКОКИНЕТИКА
ПСМ хорошо всасываются в ЖКТ, но совместный прием пищи или выраженная гипергликемия у пациента могут уменьшать скорость всасывания. Это связано с тем, что гипергликемия угнетает моторную функцию ЖКТ, что ведет к нарушению абсорбции многих ЛС. Поэтому ЛС обычно рекомендуют принимать за 30 мин до еды.
Глибенкламид имеет наименьший показатель биодоступности среди всех ПСМ, поэтому относительно недавно была разработана его так называемая микронизированная форма, обладающая улучшенными фармакокинетическими показателями (табл. 4.9).
В крови значительная часть ПСМ (90-99%) связывается с белками плазмы. Действовать ПСМ начинают спустя 2-3 ч после приема (микронизированная форма глибенкламида - через 1 ч). Несмотря на короткие периоды полувыведения, длительность действия ПСМ существенно больше, так что большинство ЛС принимают 1-2 раза в день. С одной стороны, это объясняет склонность ПСМ к распределению и кумуляции в организме, а с другой - формирование активных метаболитов. Глипизид выводится несколько быстрее остальных ПСМ, поэтому его необходимо принимать 3-4 раза в сутки, в связи с чем была разработана его новая форма - замедленного высвобождения. Она имеет осмотическую оболочку, пропускающую жидкость внутрь таблетки с постепенным высвобождением активного вещества. Эта форма глипизида получила название «гастроинтестинальная терапевтическая система», ее необходимо принимать лишь 1 раз в сутки.
Все ПСМ метаболизируют в печени, иногда с формированием активных метаболитов (глибенкламид, глимепирид). Выведение ПСМ осуществляется, как правило, через почки, но гликвидон на 95% элиминируется с желчью через кишечник, поэтому при наличии у пациента почечной недостаточности из всех ПСМ предпочтительнее это ЛС.
| ЛС | Биодоступность, % | Связывание с белками плазмы, % | Период полувыведения, ч | Продолжительность действия, ч | Путь элиминации |
|---|---|---|---|---|---|
ПСМ первого поколения |
|||||
Хлорпропамид |
90 |
90 |
36 |
24-60 |
Почечный, 100% |
ПСМ второго поколения |
|||||
Глибенкламид |
64-90 |
99 |
10-12 |
16-24 |
Почечный, 50%, печеночный, 50% |
Глибенкламид микронизированный |
90-100 |
99 |
4-10 |
12-24 |
Почечный, 50%, печеночный, 50% |
Глипизид с контролируемым высвобождением |
90 |
98-99 |
2-5 |
24 |
Почечный, 80-85% |
Гликлазид |
? |
94 |
8-11 |
12 |
Почечный, 60-70% |
Гликлазид модифицированного высвобождения |
95 |
94 |
12-20 |
24 |
Почечный, 60-70% |
Гликвидон |
95 |
98 |
1,5 |
6-8 |
Печеночный, 95% |
Глимепирид |
100 |
99 |
6-9 |
24 |
Почечный, 60% |
ПОКАЗАНИЯ
ЛС, повышающие секрецию инсулина, уже почти 60 лет с успехом применяют для лечения больных СД 2-го типа. Несмотря на наличие у большинства больных СД 2-го типа гиперинсулинемии, для преодоления имеющейся инсулинорезистентности собственного инсулина недостаточно, и необходимо медикаментозно увеличивать концентрацию гормона в крови. Хорошая компенсация СД с помощью ПСМ предупреждает и замедляет прогрессирование поздних осложнений заболевания.
ПСМ обладают наиболее выраженным гипогликемическим действием среди всех пероральных сахароснижающих препаратов: монотерапия ПСМ снижает уровень HbA1C в среднем на 1,5%. Относительная терапевтическая эффективность ПСМ второго поколения по меньшей мере в 100 раз выше по сравнению с ПСМ первого поколения, поэтому в настоящее время последние применяют редко. Лечение с помощью ПСМ рекомендуют начинать с более слабых ЛС (например, с гликлазида или глимепирида), а при неэффективности переходить на более сильные ПСМ (глибенкламид).
ПСМ второго поколения назначают, начиная с минимальных доз; при необходимости дозу постепенно увеличивают (с интервалом 1-2 нед). В табл. 4.10 показаны дозы и режим приема ПСМ. У пожилых больных следует применять ЛС с наименьшей продолжительностью действия, учитывая высокий риск гипогликемических состояний у данной категории пациентов.
| Препарат | Суточная доза, мг | Кратность приема, раз в сутки |
|---|---|---|
Глибенкламид |
2,5-20 |
1-2 |
Глибенкламид микронизированный |
1,75-14 |
1-2 |
Гликлазид |
80-320 |
1-2 |
Гликлазид модифицированного высвобождения |
30-120 |
1 |
Глипизид |
2,5-30 |
1-2 |
Глипизид с контролируемым высвобождением |
5-20 |
1 |
Глимепирид |
1-8 |
1 |
Гликвидон |
30-120 |
1-3 |
ПСМ применяют как в качестве монотерапии, так и в комбинации с другими пероральными сахароснижающими препаратами или инсулином. Следует помнить, что назначать два разных ПСМ одновременно нельзя. Монотерапия ПСМ показана больным СД 2-го типа с нормальной массой тела и сниженным уровнем C-пептида, т.е. с преобладанием недостаточной секреции инсулина.
Абсолютная недостаточность инсулина. Со временем компенсация гипергликемии на фоне ПСМ у таких пациентов может ухудшаться из-за развития абсолютной недостаточности инсулина, при этом возникают:
При истинном дефиците инсулина показано назначение той или иной схемы инсулинотерапии. Комбинированное применение ПСМ и инсулина у больных СД 2-го типа не имеет преимуществ в контроле заболевания по сравнению с монотерапией инсулином.
Комбинированная терапия. В качестве комбинированной терапии наиболее часто совместно назначают ПСМ и метформин, как правило, больным с избыточной массой тела, у которых монотерапия метформином не принесла успеха. При достижении стойкой компенсации заболевания следует попробовать снизить дозу ПСМ и вернуться к монотерапии метформином. Необходимо избегать назначения слишком высоких доз ПСМ, поскольку, с одной стороны, возрастает риск развития гипогликемических состояний, а с другой - постоянная гиперстимуляция β-клеток приводит к их истощению. Медикаментозно же вызываемая постоянная гиперинсулинемия только усиливает периферическую инсулинорезистентность; иными словами, формируется резистентность к действию ПСМ. Тогда пациента переводят на инсулинотерапию. Иногда это временная мера, и через несколько месяцев, когда чувствительность β-клеток поджелудочной железы к ПСМ восстановится, можно постепенно заменить инсулин на ПСМ. При неэффективности комбинации ПСМ и метформина или при наличии противопоказаний к назначению метформина можно использовать комбинацию ПСМ и тиазолидиндионов.
ПРОТИВОПОКАЗАНИЯ
Противопоказания к назначению ЛС, повышающих секрецию инсулина, включают СД 1-го типа, беременность, лактацию, тяжелую почечную или печеночную недостаточность.
В настоящее время большинство ПСМ по риску применения во время беременности относят к классу С; использование их у беременных женщин не рекомендуют, вместо них проводят инсулинотерапию.
ПОБОЧНЫЕ ЭФФЕКТЫ
Пациенты обычно хорошо переносят ПСМ, но возможны и побочные эффекты.
Гипогликемия - наиболее частый побочный эффект ЛС описываемой группы (особенно таких, как хлорпропамид и глибенкламид). Риск развития тяжелой гипогликемии составляет не более 1-3% для ПСМ второго поколения. Он существенно выше у лиц пожилого возраста, что объясняется большей встречаемостью в этом возрасте факторов, провоцирующих гипогликемию. К таким факторам относят:
Кроме того, следует учитывать, что с возрастом у некоторых больных масса тела снижается, и сохранение приема ПСМ в прежней дозировке может привести к гипогликемии. Лечение гипогликемии, вызванной ПСМ, проводят с помощью внутривенного введения раствора декстрозы, а учитывая длительный период действия ПСМ, могут понадобиться повторные введения в течение 24-48 ч.
Увеличение массы тела за счет увеличения секреции эндогенного инсулина на фоне приема ПСМ отмечают часто. Увеличения массы тела можно избежать при соблюдении гипокалорийной диеты.
Побочные эффекты со стороны сердечно-сосудистой системы. ПСМ первого поколения увеличивали риск смерти больных от сердечно-сосудистой патологии. Однако британское исследование United Kingdom Prospective Diabetes Study, в котором изучали ПСМ второго поколения, не выявило на фоне их приема увеличения смертности больных. Тем не менее при развитии у больных СД 2-го типа инфаркта миокарда зафиксировано статистически значимое увеличение смертности при приеме ПСМ по сравнению с больными, получавшими инсулинотерапию.
Это связано с тем, что ПСМ блокируют АТФ-зависимые K+-каналы в миокарде и коронарных сосудах, что нарушает дилатацию сосудов; ухудшается левожелудочковая функция и, как результат, формируется более обширная зона некроза.
Следовательно, при развитии у больного любого острого сердечно-сосудистого осложнения необходимо заменить лечение ПСМ на инсулинотерапию.
Побочные эффекты со стороны ЖКТ включают тошноту, рвоту, диарею, анорексию и даже холестатическую желтуху, но частота их возникновения довольно низкая.
Аллергические реакции на введение ПСМ также редки: кожную сыпь наблюдают менее чем у 1% пациентов.
Крайне редко ПСМ вызывают лейко- и тромбоцитопении, агранулоцитоз, апластическую и гемолитическую анемии.
Хлорпропамид имеет два специфических побочных эффекта.
-
Дисульфирамоподобная реакция - неприятные ощущения в виде «приливов» крови к лицу - возникают за счет ингибирования метаболизма ацетальдегида на фоне хлорпропамида после приема алкоголя.
-
Синдром неадекватной секреции АДГ. Хлорпропамид, потенцируя действие АДГ, может вызывать гипонатриемию и значительную задержку жидкости в организме.
ВЗАИМОДЕЙСТВИЯ
ЛС (в частности, β-адреноблокаторы и этанол), обладающие гипогликемическим действием, при совместном приеме с ПСМ способны провоцировать развитие гипогликемических состояний.
Некоторые ЛС вытесняют ПСМ из связи с белками плазмы, за счет чего повышается концентрация свободного ПСМ в крови и возрастает его влияние на β-клетки. К таким ЛС относят другие ПСМ (например, тиазидные диуретики), клофибрат, производные салициловой кислоты, варфарин.
Если дозу ПСМ подбирают на фоне приема средств с гипергликемизирующим действием, то резкая отмена последних может вызвать развитие гипогликемии.
Меглитиниды
Меглитиниды применяют для лечения СД 2-го типа с 1997 г. Первым ЛС стал репаглинид (производное бензойной кислоты), а чуть позже был зарегистрирован натеглинид (производное D-фенилаланина).
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Механизм действия меглитинидов, как и ПСМ, тесно связан с их действием на АТФ-зависимые K+ -каналы, но их действие опосредовано специфическими рецепторами, отличными от сульфонилмочевинных рецепторов-1. Под действием меглитинидов закрываются K+ -каналы и по описанному выше механизму возрастает чувствительность β-клеток к стимуляции глюкозой, что увеличивает секрецию инсулина в ответ на повышение уровня гликемии. Важная особенность меглитинидов заключается в том, что они восстанавливают раннюю фазу секреции инсулина, а вследствие короткого периода действия не развивается длительная гиперинсулинемия.
ФАРМАКОКИНЕТИКА
Особенность фармакокинетики меглитинидов и большое их преимущество - способность всасываться в ЖКТ за очень короткое время. Эти ЛС начинают действовать через несколько минут после приема, но продолжительность их действия гораздо меньше, чем ПСМ, - всего 3-4 ч, поэтому меглитиниды принимают при каждом приеме пищи (табл. 4.11).
Метаболизм меглитинидов осуществляет печень, а выводятся они преимущественно через кишечник, что позволяет использовать их при лечении больных СД 2-го типа с умеренным нарушением функции почек.
ПОКАЗАНИЯ
Особенности механизма действия и фармакокинетики позволили меглитинидам занять отдельную нишу в лечении СД 2-го типа. Благодаря этим особенностям группа получила свое второе название - прандиальные регуляторы гликемии. Действительно, по сравнению с ПСМ меглитиниды больше снижают постпрандиальный уровень гликемии, но в меньшей степени уменьшают уровень гликемии натощак. Меглитиниды принимают либо непосредственно перед едой, либо во время еды, либо спустя несколько минут после еды. Уровень инсулина возвращается к исходному через 3 ч после приема меглитинидов, что имитирует физиологическую секрецию инсулина на прием пищи и позволяет снизить вероятность гипогликемии в промежутках между едой. Таким образом, эти ЛС позволяют больному более гибко подходить к соблюдению режима питания. В случае пропуска приема пищи прием ЛС также пропускают, что важно для относительно молодых пациентов, ведущих активный образ жизни, так как при лечении ПСМ в этом случае возникал бы риск гипогликемии. Учитывая высокую стоимость ЛС, наиболее рациональным выглядит их назначение в комбинации с метформином тем больным, у которых монотерапия метформином не позволила получить удовлетворительную компенсацию заболевания, особенно если повышен постпранди-альный уровень гликемии. В то же время меглитиниды могут быть использованы также в качестве монотерапии (эффективность соответствует таковой для ПСМ) или в комбинации с тиазолидиндионами.
| Параметры | Репаглинид | Натеглинид |
|---|---|---|
Биодоступность, % |
56 |
73 |
Связывание с белками плазмы, % |
98 |
98 |
Период полувыведения, ч |
1 |
1,5 |
Продолжительность действия, ч |
3-4 |
3-4 |
Путь элиминации |
90% печеночный |
90% почечный |
Репаглинид более эффективно снижает уровень HbA1C по сравнению с натеглинидом. Репаглинид назначают в суточной дозе 0,5-16 мг, а натеглинид - в суточной дозе 120-480 мг. Принимают препараты 3-4 раза в сутки.
ПРОТИВОПОКАЗАНИЯ
Противопоказания к назначению меглитинидов следующие:
ПОБОЧНЫЕ ЭФФЕКТЫ
Пациенты обычно хорошо переносят меглитиниды, но возможны побочные эффекты:
ВЗАИМОДЕЙСТВИЯ
Меглитиниды следует с осторожностью использовать с препаратами, обладающими гипогликемическим действием. Репаглинид не рекомендуют комбинировать с гемфиброзилом, так как последний значительно усиливает действие данного препарата. Также действие меглитинидов существенно возрастает при совместном использовании с некоторыми хинолонами.
Бигуаниды
История применения бигуанидов в медицине уходит корнями в Средние века, когда в Европе для лечения СД использовалась Galega officinalis, или французская лилия, содержащая гуанидин - вещество, которое и оказывало определенное целебное действие. В чистом виде гуанидин слишком токсичен. В 1918-1920 гг. были разработаны первые ЛС - производные гуанидина, получившие название «бигуаниды» , один из которых использовали недолгое время в клинической практике, но вскоре запретили из-за токсического влияния на печень.
В 1957-1958 гг. вслед за внедрением первых ПСМ были предложены современные бигуаниды: фенформин¤ (фенэтилбигуанидρ ), метформин (N, N-диметилбигуанидρ ) и буформин (L-бутилбигуанидρ ).
Судьба этих трех ЛС оказалась неодинаковой. Фенформин¤ , обладающий наиболее выраженным сахароснижающим действием, начали широко применять для лечения СД 2-го типа. Но к середине 1970-х гг. было накоплено достаточное количество данных, подтверждающих относительно высокую частоту развития лактатацидоза и повышение смертности больных на фоне приема фенформина¤ . На основании этого Комитета по контролю за лекарственными веществами и пищевыми добавками (США) в 1976 г. запретил использование всех ЛС группы бигуанидов в клинической практике на территории США. Наиболее часто лактатацидоз встречался при применении фенформина¤ , поэтому от него в итоге отказались практически во всех странах мира. Буформин (в связи с относительно слабым гипогликемическим эффектом и потенциальным риском лактатацидоза) также был снят с производства. А метформин, обладающий достаточным сахароснижающим действием и редко вызывающий лактатацидоз, завоевал заслуженную популярность как в Европе, так и в России. Более того, с 1995 г. он был снова разрешен для применения на территории США.
Таким образом, в настоящее время во всех странах мира из группы бигуанидов применяют только метформин.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Механизм действия метформина (в отличие от ПСМ и меглитинидов) не связан с увеличением секреции инсулина. Метформин не изменяет и даже снижает базальный уровень секреции инсулина. Связываясь с фосфолипидами клеточных мембран [возможно, активируя циклический аденозинмонофосфат (цАМФ)-протеинкиназу], он вызывает ряд эффектов:
-
увеличивает поглощение глюкозы периферическими тканями (преимущественно скелетными мышцами). В связи с этим эффект метформина можно охарактеризовать не как гипогликемический, а как антигипергликемический. В итоге под действием метформина происходит уменьшение инсулино-резистентности;
-
снижает концентрацию свободных жирных кислот в плазме крови и улучшает другие показатели липидного спектра (в частности, снижает уровень триглицеридов);
-
замедляет всасывание глюкозы в тонком кишечнике и в некоторой степени снижает аппетит (анорексигенное действие);
-
активирует фибринолиз и снижает агрегационные свойства тромбоцитов путем подавления активности ингибитора активатора тканевого плазминогена-1.
Последние два эффекта имеют небольшое клиническое значение.
ФАРМАКОКИНЕТИКА
Всасывание метформина в основном происходит в тонком кишечнике, при смешивании с пищей процесс всасывания замедляется. В плазме крови метформин практически не связывается с белками. Период полувыведения метформина относительно небольшой - всего 1,5-4 ч. Метформин не метаболизирует в печени, а выводится с мочой в неизмененном виде. Другие фармакокинетические характеристики метформина в табл. 4.12.
| Параметр | Метформин | Пиоглитазон | Росиглитазон |
|---|---|---|---|
Биодоступность, % |
50-60 |
Нет данных |
99 |
Связывание с белками плазмы, % |
Незначительное |
99 |
99,8 |
Период полувыведения, ч |
1,5-4 |
3-7 (16-24*) |
3-4 |
Продолжительность действия, ч |
9-12 |
24 |
12-24 |
Путь элиминации |
Почечный - 90% |
Печеночный - 70-85% |
Почечный - 64% |
* Для активных метаболитов.
ПОКАЗАНИЯ
Метформин (при отсутствии противопоказаний) рекомендуют назначать всем больным с впервые выявленным СД 2-го типа в дополнение к диетотерапии и физическим нагрузкам, вне зависимости от массы тела пациентов. Высокая эффективность метформина была неоднократно доказана во многих крупных исследованиях, в том числе в исследовании United Kingdom Prospective Diabetes Study, которое показало, что строгая компенсация СД с помощью метформина предупреждает и замедляет прогрессирование поздних осложнений заболевания у больных СД 2-го типа и увеличивает продолжительность жизни больных. Монотерапия метформином снижает уровень HbA1C примерно на 1,5%, что говорит об одинаковой эффективности ПСМ и метформина. В то же время (в отличие от ПСМ) метформин не вызывает гипогликемии и увеличения массы тела.
Принципы титрации дозы метформина:
-
стартовая доза 500 мг 1-2 раза в сутки во время еды утром и/или вечером;
-
через 5-7 сут при отсутствии побочных эффектов со стороны ЖКТ по 850- 1000 мг 2 раза в сутки утром и вечером;
-
при появлении побочных эффектов со стороны ЖКТ снижают дозу до начальной и пробуют увеличить ее позднее;
-
максимальная эффективная доза обычно составляет 1000 мг 2 раза в сутки, дальнейшее увеличение дозы до 3 г/сут ненамного увеличивает эффективность препарата.
Метформин применяют в качестве комбинированной терапии вместе с ЛС практически всех других групп сахароснижающих препаратов. Наиболее часто этот препарат комбинируют с ПСМ второго поколения и иДПП-4, если невозможно достичь компенсации СД с помощью монотерапии метформином. Кроме того, у больных СД 2-го типа с ожирением, получающих инсулинотерапию, метформин иногда дополнительно назначают с целью снизить инсулинорезистентность и предотвратить дальнейшее увеличение массы тела пациента.
В настоящее время метформин рассматривают в качестве ЛС, способного предотвратить или отсрочить развитие СД 2-го типа. Использование метформина для профилактики СД 2-го типа у лиц с ожирением и НТГ изучалось в крупном рандомизированном исследовании Diabetes Prevention Program. Выяснилось, что на фоне метформина частота развития СД 2-го типа была статистически значительно ниже по сравнению с плацебо, но выше по сравнению с группой пациентов, выполнявших интенсивную программу физических упражнений и правильного питания.
Согласно рекомендациям экспертов Американской диабетической ассоциации, применение метформина (по 850 мг 2 раза в сутки) для лечения пациентов с нарушением гликемией натощак и НТГ оправданно лишь у определенной группы пациентов с наличием одного из следующих факторов:
При этом терапию метформином необходимо сочетать с немедикаментозным лечением (диета и физическая нагрузка).
СПКЯ - одна из наиболее частых причин нарушений менструального цикла и женского бесплодия. Патогенез этого заболевания напрямую связан с наличием у пациенток инсулинорезистентности. Применение ЛС, повышающих чувствительность тканей к инсулину, у женщин с СПКЯ приводит к нормализации менструальной функции, а также способствует наступлению овуляции, особенно в комбинации с кломифеном (см. «Синдром поликистозных яичников»). Следует особо отметить, что, по данным проведенного в 2005 г. исследования, в настоящее время не существует убедительных доказательств эффективности метформина в качестве ЛС для лечения ожирения или избыточной массы тела, поэтому при отсутствии сопутствующего СД 2-го типа или СПКЯ назначение метформина с целью снижения массы тела неоправданно.
ПРОТИВОПОКАЗАНИЯ
Общие противопоказания к использованию всех ЛС, повышающих чувствительность периферических тканей к инсулину:
Помимо этого, противопоказания к назначению метформина - все предрасполагающие к развитию лактатацидоза факторы:
Поскольку метформин за счет снижения инсулинорезистентности способствует наступлению овуляции у женщин с бесплодием, вызванным СПКЯ, при назначении ЛС рекомендуют использовать средства контрацепции, если женщина не планирует беременность.
В настоящее время по риску применения во время беременности метформин относят к классу В. Он не показан для лечения ГСД. Если женщина принимала этот препарат до наступления беременности, вместо него следует назначить инсулинотерапию.
ПОБОЧНЫЕ ЭФФЕКТЫ
Метформин, в отличие от ПСМ и препаратов инсулина, не вызывает гипогликемию.
Неприятные ощущения со стороны ЖКТ в начале лечения метформином предъявляют около 10-20% пациентов:
Симптомы диспепсии можно объяснить замедлением всасывания глюкозы в кишечнике, что приводит к усилению процессов брожения. К счастью, у большинства пациентов подобная симптоматика быстро проходит. Во избежание развития жалоб со стороны ЖКТ лечение метформином следует начинать с малых доз, а его прием осуществлять вместе с пищей.
Метформин нарушает всасывание витамина В12 , поэтому при длительном приеме препарата у некоторых больных уменьшается концентрация витамина В12 в сыворотке крови, хотя В12 -мегалобластная анемия вследствие использования метформина развивается исключительно редко. Всасывание комплекса, состоящего из витамина В12 и внутреннего фактора Касла, - кальций-зависимый процесс, поэтому для улучшения всасывания при снижении концентрации витамина В12 в крови следует назначить внутрь препараты кальция. Контроль уровня витамина В12 в крови рекомендуют проводить каждые 2-3 года.
Лактатацидоз - опасное для жизни состояние, вызванное накоплением лактата в организме (возникает при концентрации лактата в плазме выше 4 мэкв/л). Это наиболее грозный побочный эффект метформина, он возникает при:
При применении бигуанидов возрастает вероятность развития лактатацидоза, поскольку бигуаниды, с одной стороны, стимулируют продукцию лактата в мышцах, а с другой - подавляют глюконеогенез в печени (лактат, наряду с аланином, относят к основным субстратам этого процесса). Летальность при истинном бигуанид-индуцированном лактатацидозе может достигать 50%. Высокая частота возникновения лактатацидоза стала во многих странах причиной запрета фенформина¤ - ЛС из группы бигуанидов.
Фармакокинетика метформина имеет ряд преимуществ по сравнению с фармакокинетикой фенформина¤ : во-первых, метформин значительно меньше накапливается в мышцах, во-вторых, он имеет короткий период полувыведения. Если в физиологических условиях концентрация лактата в плазме составляет 0,5-1,5 ммоль/л, то на фоне приема метформина у пациентов без соответствующих противопоказаний она не превышает 2 ммоль/л, тогда как для развития лактатацидоза необходимо не менее 4 ммоль/л.
Вероятность лактатацидоза у больных СД 2-го типа при приеме обычных доз метформина не выше, чем при приеме других пероральных сахароснижающих препаратов. Таким образом, на фоне метформина в большинстве случаев возникает не метформин-индуцированный, а метформин-ассоциированный лактатацидоз, связанный в первую очередь с каким-либо соматическим заболеванием. Вот почему перед назначением метформина необходимо внимательно обследовать пациента, чтобы не допустить прием этого ЛС лицами, имеющими противопоказания. Развитие лактатацидоза при отсутствии каких-либо предрасполагающих факторов возможно лишь при длительной передозировке метформина.
На фоне приема метформина рекомендуют определять уровень лактата в крови 2 раза в год, хотя на практике такой частый контроль у большинства пациентов не оправдан. Повышение уровня лактата в крови нередко отмечают при выраженной физической активности, а также вследствие нарушения условий забора и преаналитической подготовки проб (например, использование жгута, длительное хранение образца крови в тепле). При внезапном появлении болей в мышцах на фоне приема метформина его следует немедленно отменить и измерить уровень лактата в крови.
ВЗАИМОДЕЙСТВИЯ
Хотя метформин сам по себе не вызывает гипогликемии, при совместном его использовании с ПСМ и инсулином риск развития гипогликемии возрастает.
Циметидин конкурентно ингибирует секрецию метформина в канальцах почек, поэтому почечный клиренс метформина при совместном приеме циметидина замедляется. При этом усиливается антигипергликемическое действие метформина; следовательно, при комбинированной терапии вместе с ПСМ или инсулином риск гипогликемии возрастает еще больше. Воздействия других блокаторов H2 -гистаминовых рецепторов на клиренс метформина не обнаружено.
Вводимые внутривенно контрастные вещества при проведении артериографии могут вызывать острую почечную недостаточность с олигурией у пациентов с начальной стадией поражения почек. Для предотвращения развития на этом фоне лактатацидоза следует отменить метформин за 48 ч до ангиографии и возобновить прием ЛС только через 48 ч после окончания процедуры.
Тиазолидиндионы
С 1997 г. в клиническую практику вошли тиазолидиндионы (глитазоны), в основе химической структуры которых лежит тиазолидиновое кольцо. Тиазолидиндионы - высокоаффинные агонисты ядерных рецепторов, активируемых пролифератором пероксисом PPAR-γ (название этого класса рецепторов связано с тем, что активация некоторых PPAR приводит к существенному увеличению числа пероксисом в печени). Первым ЛС, допущенным к использованию в клинической практике, стал троглитазон, но уже в 2000 г. его применение в ряде стран было запрещено из-за гепатотоксичности. На сегодняшний день из группы тиазолидиндионов применяют два ЛС - пиоглитазон и росиглитазон.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Механизм действия тиазолидиндионов такой же, как и метформина, и основан на повышении чувствительности периферических тканей к инсулину.
PPAR-γ регулирует транскрипцию генов, отвечающих за метаболизм углеводов и жиров. PPAR-γ преимущественно экспрессируется в жировой ткани, но он присутствует и в других тканях. Концентрация PPAR-γ в скелетных мышцах повышается у больных с избыточной массой тела и СД, причем количество PPAR-γ положительно коррелирует с концентрацией инсулина в плазме крови.
Вероятно, снижение инсулинорезистентности на фоне тиазолидиндионов происходит за счет повышения синтеза транспортеров глюкозы; при этом увеличивается транспорт глюкозы внутрь адипоцитов и миоцитов, где активируются процессы синтеза гликогена и гликолиза. Тиазолидиндионы действуют лишь при наличии инсулина - они, как и метформин, помогают инсулину оказывать действие на ткани организма. Следует отметить, что тиазолидиндионы, по сравнению с метформином, в значительно большей степени снижают инсулинорезистентность тканей, а глюконеогенез в печени они подавляют незначительно.
ФАРМАКОКИНЕТИКА
Тиазолидиндионы хорошо всасываются в ЖКТ и почти полностью связываются с белками плазмы (см. табл. 4.11). Максимальную концентрацию пиоглитазон в крови достигает через 2-4 ч, росиглитазон - уже через 1 ч. Тиазолидиндионы метаболизируются в печени, при этом у пиоглитазона происходит формирование активных метаболитов, что обеспечивает большую продолжительность его действия. Несмотря на то что росиглитазон выводится преимущественно почками, при легкой и средней степени почечной недостаточности ЛС можно назначать без корректировки дозы.
ПОКАЗАНИЯ
Применение тиазолидиндионов для лечения СД 2-го типа наиболее рационально в сочетании с метформином, если монотерапия метформином не принесла успеха, либо в сочетании с ПСМ, если метформин не эффективен или противопоказан у данного пациента (например, в связи с почечной недостаточностью). Комбинированная терапия тиазолидиндионами и ИСД (ИДД) также позволяет улучшить контроль заболевания (уровень HbA1C может дополнительно снижаться более чем на 1%), причем при меньших дозах инсулина у тех больных, которые получали ранее монотерапию большими дозами инсулина (более 30 ЕД/сут). В то же время при совместном использовании тиазолидиндионов и инсулина существенно повышается вероятность задержки жидкости в организме (см. подраздел «Побочные эффекты»). В связи с этим данную комбинацию ЛС следует с осторожностью назначать пациентам из группы риска по развитию сердечной недостаточности.
Применение тиазолидиндионов для профилактики СД 2-го типа у пациентов из группы высокого риска на сегодняшний день - дискуссионный вопрос. По данным рандомизированного исследования Troglitazone in Prevention of Diabetes, прием троглитазона в значительной степени снижал частоту развития СД 2-го типа по сравнению с плацебо у женщин, имевших ГСД в анамнезе. Хотя троглитазон к настоящему моменту уже не рекомендован к использованию, результаты указанного исследования позволяют предположить, что и другие тиазолидиндионы способны предотвращать развитие СД 2-го типа. Действительно, в крупном исследовании Diabetes Reduction Assessment with Ramipril and Rosiglitazone Medication прием росиглитазона снижал риск развития СД 2-го типа. Тем не менее на сегодняшний день тиазолидиндионы не показаны для широкого использования с целью профилактики данного заболевания.
Максимальная суточная доза пиоглитазона 15-45 мг, назначают 1 раз в сутки. Росиглитазон применяют 1-2 раза в сутки в дозе 2-8 мг/сут.
ПРОТИВОПОКАЗАНИЯ
Общие противопоказания к использованию всех ЛС, повышающих чувствительность периферических тканей к инсулину, - СД 1-го типа, беременность и лактация. Тиазолидиндионы противопоказаны при сердечной недостаточности III и IV класса, а также при уровне аланинаминотрансферазы (АЛТ) выше верхней границы нормы в 2,5 раза.
В настоящее время по риску применения во время беременности пиоглитазон и росиглитазон относят к классу C. Использование их для лечения ГСД не рекомендуют. Если женщина принимала эти ЛС до наступления беременности, вместо них следует назначить инсулинотерапию.
ПОБОЧНЫЕ ЭФФЕКТЫ
Тиазолидиндионы, в отличие от ПСМ и инсулина, не вызывают гипогликемию, их применение сопряжено с развитием иных побочных эффектов.
Гепатотоксичность. Именно из-за гепатотоксичности троглитазон (первое ЛС из этой группы) был снят в США с производства. Другие тиазолидиндионы не обладают гепатотоксичностью, так как в контролируемых клинических исследованиях частота повышения уровня АЛТ на фоне терапии тиазолидиндионами не отличалась от таковой на фоне приема других пероральных сахароснижающих препаратов. При этом гепатотоксичность троглитазона связывают с наличием в его составе токоферольного кольца, которое отсутствует у росиглитазона и пиоглитазона.
Также возможно развитие:
Сообщений о гепатотоксичности пиоглитазона на момент публикации в доступных источниках не обнаружено.
В связи с вышесказанным перед назначением тиазолидиндионов следует оценить функцию печени. При наличии клинических признаков активного заболевания печени или при уровне АЛТ выше верхней границы нормы в 2,5 раза от использования тиазолидиндионов следует воздержаться.
В первый год приема тиазолидиндионов необходимо регулярно (обычно каждые 2-3 мес) определять уровень АЛТ в сыворотке крови. При небольшом исходном повышении уровня АЛТ (до 2,5 раза от верхней границы нормы) следует контролировать уровень АЛТ еще чаще.
Если в процессе лечения уровень АЛТ становится в 3 раза выше верхней границы нормы, рекомендуют повторить анализ и при аналогичном результате прекратить прием ЛС. При появлении желтухи ЛС также отменяют.
Увеличение массы тела - побочный эффект, который отсутствует у метформина (в отличие от тиазолидиндионов). Данное явление имеет дозозависимый и времязависимый характер. Следует отметить, что увеличение массы тела наблюдают как на фоне монотерапии тиазолидиндионами, так и при их сочетании с ПСМ или инсулином, причем в последнем случае масса тела увеличивается наиболее существенно. Природа этого феномена не вполне ясна. С одной стороны, компенсация СД устраняет глюкозурию и увеличивает реальную калорийность пищи, что закономерно обусловливает повышение массы тела. С другой стороны, происходит пролиферация новых адипоцитов, что вызывает перераспределение жировой ткани в сторону увеличения подкожного «депо».
Задержка жидкости в организме - частый побочный эффект тиазолидиндионов и, по-видимому, наиболее весомая причина увеличения массы тела. Задержка жидкости способствует возникновению не только увеличения массы тела, но и периферических отеков, сердечной недостаточности, а также анемии вследствие гемодилюции.
Отеки стоп на фоне монотерапии тиазолидиндионами возникают у 3-5% больных. При назначении этих ЛС в комбинации с другими пероральными сахароснижающими препаратами частота периферических отеков еще больше возрастает. При одновременном назначении тиазолидиндионов с инсулином частота периферических отеков составляет примерно 13-16%. При развитии отеков стоп на фоне терапии тиазолидиндионами следует в первую очередь исключить сердечную недостаточность и другие возможные причины возникновения отеков (нефротический синдром, терапию дигидропиридиновыми антагонистами кальция). В случае необходимости для лечения отеков стоп, вызванных тиазолидиндионами, используют диуретики.
Сердечная недостаточность на фоне монотерапии тиазолидиндионами развивается менее чем у 1% пациентов. В то же время при добавлении тиазолидиндионов к инсулинотерапии частота сердечной недостаточности возрастает до 2-3% по сравнению с 1% на фоне монотерапии инсулином. При развитии сердечной недостаточности на фоне терапии тиазолидиндионами следует тщательно обдумать необходимость их дальнейшего применения у данного пациента. Если у пациента ранее имелась дисфункция левого желудочка, тиазолидиндионы отменяют в обязательном порядке.
Росиглитазон и пиоглитазон в соответствующих дозах примерно в одинаковой степени вызывают упомянутые побочные эффекты, хотя прямых сравнительных исследований не проводилось.
При наличии факторов риска развития сердечной недостаточности стартовую дозу ЛС уменьшают (росиглитазона до 4 мг, пиоглитазона до 15 мг). Увеличивать дозу до оптимальной необходимо под строгим контролем для выявления возможных признаков сердечной недостаточности.
Факторы риска развития сердечной недостаточности:
Если у больного сердечная недостаточность функциональных I-II класса, лечение тиазолидиндионами следует начинать с минимальных доз: 2 мг росиглитазона и 15 мг пиоглитазона.
В 2007 г. появились данные о том, что росиглитазон, возможно, повышает риск инфаркта миокарда, сердечно-сосудистой смерти и переломов костей (у женщин), поэтому он был запрещен в Европе и ограничена его продажа в США, но эти факты требуют дальнейшего изучения.
ВЗАИМОДЕЙСТВИЯ
Эффективность тиазолидиндионов существенно возрастает при совместном использовании с гемфиброзилом и некоторыми хинолонами.
Ингибиторы α-глюкозидаз
Разработка данной группы ЛС началась в конце 1960-х гг. Идея состояла в том, чтобы блокировать деятельность особых ферментов кишечника - α-глюкозидаз. Дисахариды и олигосахариды в кишечнике не всасываются, но под действием α-глюкозидаз расщепляются там до всасывающихся моносахаридов. Первые ингибиторы α-глюкозидаз, полученные из пшеничной муки, действовали только на панкреатическую α-амилазу, поэтому клинический эффект был выражен слабо. Спустя некоторое время из культуры Actinoplanes utanhensis был выделен псевдотетрасахарид, который впоследствии был назван акарбозой. Вторым ЛС, внедренным в широкую клиническую практику, стал миглитол - производное дезоксинойримицина, которое по своей структуре можно отнести к моносахаридам.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Механизм действия ингибиторов α-глюкозидаз в первую очередь связан с их влиянием на ферменты, расположенные в «щеточной каемке» энтероцитов. Акарбоза и миглитол обратимо и конкурентно ингибируют α-глюкозидазы: глюкамилазу, сукразу, декстриназу, мальтазу и лишь в небольшой степени α-амилазу (акарбозу) и лактазу (миглитол). Вследствие фармакокинетических особенностей данных ЛС действие их в основном происходит в верхней части тонкого кишечника. В дистальной части тонкого кишечника способность ингибировать α-глюкозидазы ослабляется, поэтому непереваренные олиго- и дисахариды все же расщепляются на моносахариды и всасываются внутрь энтероцитов. Таким образом, ингибиторы α-глюкозидаз замедляют процессы ферментирования сложных углеводов и, как следствие, уменьшают скорость всасывания продуктов ферментирования (моносахаридов). Соответственно не происходит резкого подъема уровня гликемии после еды. На процесс всасывания простых углеводов (глюкозы, фруктозы) ни акарбоза, ни миглитол не оказывают влияния, поэтому антигипергликемическое действие ингибиторов α-глюкозидаз проявляется лишь при преимущественном употреблении в пищу сложных углеводов (продуктов, содержащих крахмал, декстрины, дисахариды).
ФАРМАКОКИНЕТИКА
Ингибиторы α-глюкозидаз действуют непосредственно в тонком кишечнике. Лишь 2% от поглощенной дозы акарбозы всасывается и попадает в системный кровоток, а основную часть акарбозы в итоге расщепляют населяющие тонкий кишечник микроорганизмы. Миглитол, напротив, полностью всасывается в проксимальной части тонкого кишечника. Периоды полувыведения миглитола и акарбозы из плазмы крови - около 2 ч, элиминация осуществляется почками.
ПОКАЗАНИЯ
ЛС, нарушающие всасывание углеводов в кишечнике, применяют для лечения СД 2-го типа (в основном в комбинации с другими пероральными сахароснижающими препаратами). При этом используют способность ингибиторов α-глюкозидаз эффективно снижать постпрандиальный уровень гликемии, а коррекции уровня гликемии натощак обычно достигают с помощью ПСМ или метформина. На фоне приема ингибиторов α-глюкозидаз фармакокинетика ПСМ и метформина не изменяется. Ингибиторы α-глюкозидаз также можно сочетать с инсулинотерапией.
Схема титрации дозы ингибиторов α-глюкозидаз:
Обычная суточная доза акарбозы 150-300 мг, миглитола - 75-300; оба препарата применяют 3 раза в сутки.
Всплеск интереса к данной группе ЛС произошел после опубликования результатов двойного слепого плацебоконтролированного исследования STOP-NIDDM, в котором акарбоза существенно снижала риск развития СД 2-го типа у лиц с избыточной массой тела и НТГ.
В инструкции к акарбозе есть показание - для профилактики СД 2-го типа.
ПРОТИВОПОКАЗАНИЯ
Противопоказания к назначению ЛС, блокирующих α-глюкозидазы, включают беременность, лактацию, хронические заболевания кишечника, острые и хронические гепатиты и панкреатиты, а также возраст моложе 18 лет.
ПОБОЧНЫЕ ЭФФЕКТЫ
Побочные эффекты ингибиторов α-глюкозидаз нельзя назвать опасными, тем не менее они частые причины отмены этих ЛС.
В результате действия α-глюкозидаз в толстый кишечник поступает значительное количество углеводов. Здесь они подвергаются процессам брожения с образованием большого количества газов. Вследствие этого у пациентов часто возникают метеоризм и диарея. Выраженность побочных эффектов можно уменьшить, если начинать терапию с небольших доз и увеличивать дозу постепенно. ЛС необходимо принимать не разжевывая, с небольшим количеством жидкости, непосредственно перед или во время еды.
Гипогликемия на фоне терапии ингибиторами α-глюкозидаз не развивается, но если она возникает по другой причине (например, вследствие передозировки ПСМ), то ЛС из данной группы могут существенно замедлить всасывание углеводов, принимаемых внутрь для коррекции гипогликемии. Иными словами, несмотря на прием углеводов (сахара, мучных изделий) внутрь, гипогликемия может усугубляться. В такой ситуации для коррекции гипогликемии пациенту следует использовать продукты, содержащие чистую глюкозу [раствор или таблетки декстрозы (глюкозы♠ )].
Повышение активности аминотрансфераз. Активность аспартатамино-трансферазы (АСТ) и АЛТ иногда увеличивается у пациентов, принимающих акарбозу (особенно в высоких дозах), не совсем ясно, по какой причине. В связи с этим в первый год приема ингибиторов α-глюкозидаз необходимо регулярно (обычно каждые 3 мес) определять активность АЛТ и АСТ в сыворотке крови. При повышении активности ферментов необходимо снизить дозу ЛС. При стойком повышении активности АЛТ и АСТ следует решить вопрос о целесообразности дальнейшего продолжения приема ингибиторов α-глюкозидаз.
ВЗАИМОДЕЙСТВИЯ
Эффективность ингибиторов α-глюкозидаз может снижаться при совместном назначении с ЛС, содержащими пищеварительные ферменты.
Препараты, основанные на действии инкретинов
болочку, пропускающую жидкость внутрь таблетки с постепенным высвобождением активного вещества. Эта форма глипизида получила название «гастроинтестинальная терапевтическая система», ее необходимо принимать лишь 1 раз в сутки. Инкретины - это гормоны, вызывающие стимуляцию секреции инсулина после перорального приема декстрозы (глюкозы♠ ). После приема внутрь раствора декстрозы (глюкозы♠ ) или пищи секреция инсулина увеличивается в большей степени, чем после внутривенного введения соответствующего количества декстрозы (глюкозы♠ ). Этот феномен обозначается как «инкретиновый эффект» и обусловлен действием гормонов-инкретинов.
Двумя основными инкреторными гормонами у людей являются глюкозозависимый инсулинотропный полипептид (ГИП) и ГПП-1. ГИП - это белок, состоящий из 42 аминокислот, отщепленный от более крупного белка (про-ГИП) и секретируемый эндокринными К-клетками, преимущественно содержащимися в проксимальной части ЖКТ - двенадцатиперстной кишке и проксимальном отделе тощей кишки. ГПП-1 - это белок, состоящий из 30 или 31 аминокислоты, отщепленный от более крупного белка (проглюкагона) и секретируемый L-клетками, которые по большей части располагаются в дистальной части ЖКТ - подвздошной и толстой кишке.
Регуляторное действие ГПП-1 и ГИП на уровень глюкозы осуществляется через связывание и активацию соответствующих рецепторов (ГПП-1R-1р и ГИП-R), расположенных в различных тканях, включая α- и β-клетки островков поджелудочной железы (табл. 4.13). Натощак концентрации ГПП-1 и ГИП в плазме низкие, однако после приема пищи секреция ГПП-1 и ГИП быстро возрастает.
| Органы и системы | Эффекты ГПП-1 |
|---|---|
Поджелудочная железа |
Усиливает глюкозостимулированную секрецию инсулина. Стимулирует транскрипцию гена инсулина матричной рибонуклеиновой кислоты (РНК). Ингибирует секрецию глюкагона. Ингибирует секрецию соматостатина. Регулирует экспрессию и активность АТФ-зависимых калиевых каналов β-клеток. Усиливает ответную реакцию β-клеток на глюкозу. Вызывает неогенез и пролиферацию β-клеток |
Желудочно-кишечная система |
Ингибирует опустошение желудка. Ингибирует кислотную секрецию желудка |
Сердечно-сосудистая система |
Усиливает скорость сердечных сокращений |
Центральная нервная система (ЦНС) |
Подавляет поступление пищи и воды. Стимулирует секрецию рилизинг-гормона-ЛГ из гипоталамуса. Стимулирует секрецию ТТГ, ЛГ, кортикостероидов |
Мышцы, жировая ткань и печень |
Усиливает инсулин-стимулированный метаболизм глюкозы в адипоцитах. Стимулирует включение глюкозы в гликоген, в гепатоциты и скелетные мышцы. Является посредником в обеспечении гепатопортальной чувствительности к глюкозе |
Щитовидная железа, легкие, почки |
Стимулирует выделение кальцитонина из щитовидной железы. Усиливает мукозную секрецию и релаксацию легочных мышц. Способствует диурезу и экскреции натрия в почках |
После высвобождения ГПП-1 (7-36) и ГИП (1-42) быстро метаболизируются ферментом дипептидилпептидазой-4 (ДПП), который располагается на мембранах щеточной каемки кишечного и почечного эпителия, на поверхности капилляров и присутствует в кровотоке в растворимой форме. ДПП-4 осуществляет отщепление двух аминокислот от интактного пептида, в результате чего образуются два укороченных пептида ГПП-1 (9-36) и ГИП (3-42), которые выводятся из организма главным образом почками. Период полувыведения инкретинов из плазмы короткий (приблизительно 2 мин для интактного ГПП-1 и до 5 мин - для ГИП). После быстрой деградации метаболиты ГПП-1 и ГИП выводятся из организма через почки (рис. 4.4).
Наибольше клиническое значение нашли эффекты ГПП-1. Для воспроизведения его действия ГПП-1 было предложено два пути.
-
Синтез препарата, способного воздействовать на рецепторы ГПП-1 в организме человека и воспроизводить все его эффекты. При этом желательно создать молекулу, устойчивую к действию фермента ДПП-4. Такие препараты были созданы и получили название аГПП-1.
-
Пролонгация действия эндогенного ГПП-1 путем создания препаратов, ингибирующих активность фермента ДПП-4. Эти препараты называются иДПП-4.
Обе группы препаратов вошли в широкую клиническую практику около 10 лет назад.
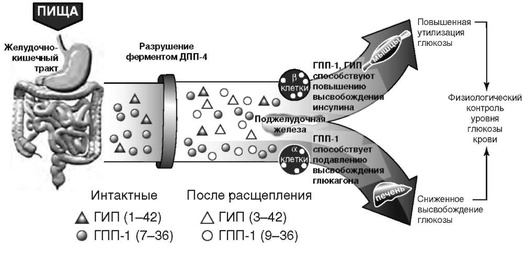
Агонисты рецепторов глюкагоноподобного пептида-1
У здоровых людей «инкретиновый эффект» составляет от 20 до 60% постпрандиальной секреции инсулина. В то же время у больных СД 2-го типа он может быть существенно ослаблен, у таких пациентов инсулиновый ответ после перорального назначения декстрозы (глюкозы♠ ) снижен и отсрочен во времени. Наиболее важным и хорошо изученным представителем инкретинов является ГПП-1.
ГПП-1 является посттрансляционным продуктом гена проглюкагона, членом суперсемейства глюкагона, в которое входят такие пептидные гормоны, как глюкагон, ГПП-1, ГПП-2, желудочный ингибирующий пептид и экзендин-4.
В настоящее время в клинической практике для лечения СД 2-го типа используются агонисты рецепторов ГПП-1. В начале 90-х гг. прошлого столетия исследователи обнаружили, что действие эксендина-4 - пептида, выделенного из слюнных желез ящерицы Gilamonster, - очень похоже на действие ГПП-1, вырабатываемого эндокринными клетками пищеварительного тракта человеческого организма. Но в сравнении с ГПП-1 эксендин-4 обладает существенно большей продолжительностью действия. Это открытие послужило основой для создания синтетического аналога эксендина-4 для лечения больных СД 2-го типа, названного эксенатид. В дальнейшем был создан аналог человеческого ГПП-1 - лираглутид. Он гомологичен нативному ГПП-1 на 97%. Третьим препаратом, зарегистрированным в России, является ликсисенатид. Все три препарата предназначены для ежедневного применения. В мире зарегистрированы препараты этой группы пролонгированного действия - албиглутидρ и дулаглутидρ , которые вводятся 1 раз в неделю.
Терапия агонистами рецепторов ГПП-1 является патогенетической, так как направлена на восстановление естественных физиологических механизмов регуляции уровня глюкозы. В ряде исследований было показано, что агонист рецепторов ГПП-1 вызывал снижение уровня глюкагона в циркуляции крови, увеличение массы β-клеток поджелудочной железы, улучшение секреции инсулина, уменьшение аппетита наряду с достижением хорошего гликемического контроля.
Относительно неудобным можно считать подкожный путь введении препаратов. Препарат применяется в фиксированных дозах, не требует сложного режима титрования и специального обучения пациентов, что особенно актуально для пожилых пациентов с СД 2-го типа.
Важным преимуществом препаратов является то, что благодаря физиологическому механизму регуляции секреции инсулина он сам по себе не вызывает гипогликемии, она возможна только при сочетанном применении с другими сахароснижающими препаратами.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Препараты, относящиеся к группе агонистов рецепторов ГПП-1, представляют собой генно-инженерные пептиды, гомологичные ГПП-1 человека и обладающие всем спектром действия, характерным для этого гормона ЖКТ:
Глюкозозависимый механизм действия аГПП-1 позволяет контролировать уровень гликемии (как натощак, так и постпрандиальный) без риска развития гипогликемических состояний, поскольку при снижении гликемии до уровня 4,5 ммоль/л прекращается инсулин-стимулирующее и глюкагон-подавляющее действие препарата из группы аГПП-1.
Экспериментальные данные свидетельствуют о том, что лечение аГПП-1 приводит к сохранению массы β-клеток за счет их неогенеза из клеток протоков поджелудочной железы.
ПОКАЗАНИЯ
СД 2-го типа в качестве монотерапии или дополнительной терапии к метформину, ПСМ, тиазолидиндионам, комбинации метформина и ПСМ или метформина и тиазолидиндионов, а также в комбинации с базальным инсулином в случае недостижения адекватного гликемического контроля.
ПРОТИВОПОКАЗАНИЯ
Гиперчувствительность, СД 1-го типа, ДКА, тяжелая ХБП (СКФ менее 30 мл/мин, 1,73 м2 ), тяжелые заболеваний ЖКТ с сопутствующим гастропарезом, беременность, период лактации, возраст до 18 лет (безопасность и эффективность не установлены).
ПОБОЧНЫЕ ЭФФЕКТЫ
Достаточно частым побочным эффектом применения являются желудочно-кишечные нарушения (тошнота, рвота, диарея, снижение аппетита), гипогликемия (в комбинации с ПСМ). Иногда возникают ощущение дрожи, головокружение, головная боль, слабость, гастроэзофагеальный рефлюкс, гипергидроз, кожная реакция в месте инъекции, абдоминальная боль, вздутие живота, отрыжка, запор, нарушение вкусовых ощущений, метеоризм. Крайне редко бывают анафилактические реакции.
На фоне терапии возможно появление антител к эксенатиду, что, однако, не влияет на частоту и тип регистрируемых побочных эффектов. Образование антител к лираглутиду очень мало.
Сообщалось о нескольких случаях повышения времени свертывания крови, иногда сопровождавшегося кровотечениями, при одновременном применении с варфарином.
Связь с развитием панкреатита на сегодняшний день не является доказанной. Более подробная характеристика зарегистрированных в РФ агонистов ГПП-1 представлена ниже.
ЭКСЕНАТИД
Представляет собой синтетический аналог эксендина-4.
Фармакокинетика
Быстро всасывается после подкожного введения, время достижения максимальной концентрации - 2,1 ч, максимальной концентрации - 211 пг/мл, системной экспозиции - 1036 пг×ч/мл после подкожного введения 10 мкг. Измеряемые концентрации эксенатида определяются приблизительно в течение 10 ч после введения. Системная экспозиция возрастает пропорционально увеличению дозы с 5 до 10 мкг, при этом пропорционального возрастания максимальной концентрации не наблюдается. Сходная экспозиция наблюдается при подкожном введении эксенатида в область живота, бедра или предплечья. Объем распределения 28,3 л.
Выводится преимущественно за счет клубочковой фильтрации с последующим протеолитическим распадом. Клиренс 9,1 л/ч, период полувыведения 2,4 ч, вне зависимости от дозы.
Форма выпуска и режим дозирования
Эксенатид выпускается в виде раствора для подкожного введения, упакованного в предварительно заполненную шприц-ручку. Препарат выпускается в двух дозировках (5 и 10 мкг), поэтому предлагаются 2 разные шприц-ручки, на которых указаны соответствующие концентрации эксенатида. Путь введения - подкожно в область бедра, живота или предплечья. Начальная доза 5 мкг 2 раза в сутки в любое время в пределах 1 ч перед утренним и вечерним приемом пищи. Не следует назначать препарат после приема пищи. В случае пропуска инъекции препарата лечение продолжают без изменения дозы. Через 1 мес после начала лечения дозу препарата можно увеличить до 10 мкг 2 раза в сутки. При одновременном применении с метформином, тиазолидиндионом или с комбинацией этих двух препаратов их исходная доза не изменяется. В случае комбинации с ПСМ может потребоваться снижение дозы последнего для снижения риска гипогликемии.
Взаимодействия
При применении пероральных ЛС, требующих быстрого всасывания в ЖКТ, необходимо учитывать, что препарат может замедлять опорожнение желудка. Препараты, действие которых зависит от их пороговой концентрации (в т.ч. антибиотики), рекомендуется применять не менее чем за 1 ч до введения эксенатида; если эти препараты необходимо принимать с пищей, следует принимать их во время тех приемов пищи, когда эксенатид не вводится.
Повышает максимальную концентрацию дигоксина на 17%, время достижения максимальной концентрации на 2,5 ч, при этом системная экспозиция не изменяется.
Снижает системную экспозицию и максимальную концентрацию ловастатина приблизительно на 40 и 28% соответственно, увеличивает время достижения максимальной концентрации на 4 ч.
Увеличивает ТCmax лизиноприла на 2 ч (при этом не наблюдалось изменений показателей среднесуточного систолического и диастолического АД).
При введении варфарина через 30 мин после эксенатида время достижения максимальной концентрации варфарина увеличивалось примерно на 2 ч, однако клинически значимого влияния на максимальную концентрацию или системную экспозицию не наблюдалось.
Новая форма эксенатида - эксенатид-LARρ
Разработана и зарегистрирована (не в России) пролонгированная форма эксенатида под названием «эксенатид-LARρ », который вводится 1 раз в неделю. Эксенатид-LARρ может обладать более выраженным сахароснижающим действием по сравнению с обычным эксенатидом.
ЛИРАГЛУТИД
Лираглутид представляет собой первый препарат из группы аналогов человеческого ГПП-1. Он синтезирован генно-инженерным способом в результате модификации нативного ГПП-1 путем замены одной аминокислоты (аргинин на лизин) в позиции 34 и добавлению к лизину в 26-й позиции пальмитиновой кислоты (С16).
В результате аминокислотный состав лираглутида на 97% гомологичен человеческому ГПП-1, тогда как эксенатид имеет гомологичность только на 53%. Более того, такая модификация обеспечила резистентность лираглутида к расщеплению под действием фермента ДПП-4, а также его способность связываться с альбумином плазмы и образовывать мицелоподобные агрегаты в подкожно-жировой клетчатке.
Фармакокинетика
Всасывание лираглутида после подкожного введения происходит медленно, максимальное время в плазме составляет 8-12 ч после введения дозы препарата. Лираглутид в значительной степени связывается с белками плазмы крови (>98%). Лираглутид метаболизируется с образованием двух метаболитов, которые затем метаболизируются эндогенно, подобно крупным белкам, без привлечения какого-либо специфического органа в качестве пути выведения. Метаболиты лираглутида лишь в незначительном количестве (6 и 5% соответственно) выводятся почками или через кишечник. Период его полувыведения составляет 10-14 ч, что обеспечивает его стабильную концентрацию при однократном введении в сутки уже после трех последовательных инъекций.
Пол, возраст, ИМТ не оказывают клинически значимого эффекта на фармакокинетические свойства лираглутида.
Форма выпуска и режим дозирования
Лираглутид выпускается в виде раствора для подкожного введения, в предварительно заполненной шприц-ручке. Каждая ручка может быть предназначена для введения 15 доз по 1,2 мг или 10 доз по 1,8 мг, а также имеется возможность вводить 0,6 мг в начале лечения. Ручка должна храниться в холодильнике (2-8 °С); после первого использования она может в течение месяца находиться при комнатной температуре (ниже 25 °С); ручку нельзя замораживать.
Лираглутид назначается один раз в сутки подкожно в область живота, бедра или плеча в любое время дня. Начальная доза лираглутида составляет 0,6 мг/ сут в течение недели для оптимальной переносимости препарата. Со второй недели дозу необходимо увеличить до 1,2 мг/сут. У некоторых пациентов может возникнуть необходимость в применении максимальной дозы 1,8 мг, в случае отсутствия достижения целей гликемического контроля при использовании дозы 1,2 мг. Благодаря глюкозозависимому действию лираглутида для подбора его дозы не требуется частого мониторинга уровня гликемии. Такой самоконтроль гликемии может понадобиться при назначении лираглутида в комбинации с ПСМ.
Взаимодействия
Лираглутид показал очень низкую способность к фармакокинетическому взаимодействию с ЛС, обусловленному метаболизмом в системе цитохрома Р450 (CYP), а также связыванием с белками плазмы. Небольшая задержка в опорожнении желудка при применении лираглутида может оказывать влияние на всасывание сопутствующих пероральных ЛС. Исследования лекарственного взаимодействия не показали какого-либо клинически значимого замедления всасывания этих препаратов. У нескольких пациентов, получавших лечение лираглутидом, отмечалось минимум по одному эпизоду острой диареи. Диарея может оказывать влияние на всасывание пероральных ЛС, которые используются одновременно с лираглутидом. Вещества, добавленные к лираглутиду, могут вызвать его деградацию, поэтому препарат нельзя смешивать с другими ЛС, в том числе с инфузионными растворами.
Новая форма лираглутида
Столь высокая эффективность лираглутида в отношении снижения массы тела позволила зарегистрировать (не в России) лираглутид в дозе 3 мг для лечения ожирения.
ЛИКСИСЕНАТИД
Фармакокинетика
После подкожного введения ликсисенатида скорость его абсорбции является высокой и не зависит от введенной дозы. Медиана времени достижения максимальной концентрации ликсисенатида в крови составляет 1-3,5 ч. Ликсисенатид имеет умеренную степень связи с белками крови у человека (55%). Независимо от вводимой дозы объем распределения после подкожного введения ликсисенатида составляет 90-140 л после однократного введения и 90-120 л при повторном введении. Ликсисенатид выводится с помощью гломерулярной фильтрации с последующей канальцевой реабсорбцией и метаболической деградацией, приводящей к образованию более мелких пептидов и аминокислот, которые повторно вовлекаются в белковый обмен.
После повторного введения среднее значение периода полувыведения обычно находилось в диапазоне 1,5-4,5 ч, среднее значение клиренса находилось в диапазоне 20-67 л/ч.
Форма выпуска и режим дозирования
Раствор для подкожного введения 0,05 мг/мл (10 мкг/доза) и 0,1 мг/мл (20 мкг/доза), по 3 мл препарата в предварительно заполненной шприц-ручке.
Препарат вводится 1 раз в сутки в пределах 1 ч до первого в течение дня приема пищи или в пределах 1 ч до вечернего приема пищи. В случае пропуска введения очередной дозы ее следует ввести в пределах 1 ч до следующего приема пищи. Ликсисенатид вводится подкожно в область бедра, брюшной стенки или плеча. Начальная доза составляет 10 мкг 1 раз в сутки в течение 14 дней. Затем доза препарата должна быть увеличена до 20 мкг 1 раз в сутки. Эта доза является поддерживающей. Когда препарат добавляется к уже проводимой терапии метформином, прием метформина может быть продолжен без изменения его дозы. Когда препарат добавляется к уже проводимой терапии ПСМ или базальным инсулином, для уменьшения риска развития гипогликемии можно рассмотреть вопрос о снижении дозы ПСМ или инсулина. Применение ликсисенатида не требует специального мониторинга концентрации глюкозы в крови. Однако при применении его в комбинации с ПСМ или базальным инсулином может потребоваться дополнительный самоконтроль гликемии.
До использования шприц-ручка должна храниться в холодильнике при температуре 2-8 °С. После первого использования шприц-ручка может храниться при температуре не выше 30 °С не более 14 дней. После каждого использования шприц-ручку следует закрывать колпачком для того, чтобы защитить ее от воздействия света. Шприц-ручку не следует хранить с присоединенной иглой.
Взаимодействия
Ликсисенатид является пептидом и не метаболизируется с помощью изоферментов цитохрома P450. В in vitro -исследованиях, проведенных у человека, ликсисенатид не нарушал активности протестированных изоферментов цитохрома P450 или транспортеров. Задержка опорожнения желудка при применении ликсисенатида может повлиять на скорость абсорбции ЛС, принимаемых внутрь. Для принимаемых внутрь ЛС, эффективность которых особенно зависит от пороговых концентраций, пациентам следует рекомендовать принимать эти ЛС как минимум за 1 ч до или спустя 11 ч после инъекции ликсисенатида.
Ингибиторы дипептидилпептидазы-4
На сегодняшний день зарегистрированы на территории РФ и внедрены в клиническую практику несколько иДПП-4: ситаглиптин, вилдаглиптин, саксаглиптин, линаглиптин и алоглиптин.
Препараты предназначены для перорального применения однократно или два раза в сутки.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
иДПП-4 ингибируют активность фермента ДПП-4 на поверхности эндотелиальных клеток капилляров слизистой кишечника, вследствие чего эндогенные гормоны-инкретины (ГПП-1 и ГИП) не разрушаются и сохраняют свою активность в течение 12-24 ч. Их концентрация возрастает приблизительно в 2-3 раза, особенно после пищевой нагрузки, т.е. восстанавливается физиологическая или чуть выше физиологической концентрация инкретинов, в частности ГПП-1. Далее наблюдаются все основные эффекты, которые свойственны ГПП-1:
В отличие от аГПП-1, не вызывают замедления опорожнения желудка и оказывают нейтральное влияние на массу тела. Есть экспериментальные данные об увеличении массы β-клеток на фоне лечения иДПП-4.
ПОКАЗАНИЯ
СД 2-го типа в качестве монотерапии или дополнительной терапии к метформину, ПСМ, тиазолидиндионам, комбинации метформина и ПСМ или метфор-мина и тиазолидиндиона, а также в комбинации с базальным инсулином в случае недостижения адекватного гликемического контроля.
ПРОТИВОПОКАЗАНИЯ
Гиперчувствительность, СД 1-го типа, ДКА, беременность, период лактации, возраст до 18 лет (безопасность и эффективность не установлены). иДПП-4 разрешены к применению на на всех стадиях ХБП, включая терминальную, с соответствующим снижением дозы (линаглиптин без снижения дозы). С осторожностью при тяжелой печеночной недостаточности (кроме саксаглиптина, линаглиптина). Коррекция дозы у пожилых лиц не требуется, за исключением случаев, сочетающихся со снижением СКФ. Достаточных данных о применении препарата у лиц моложе 18 лет и в педиатрической практике нет. Поэтому у данной категории использование препарата пока не рекомендуется.
ПОБОЧНЫЕ ЭФФЕКТЫ
В большинстве исследований иДПП-4 имели профиль переносимости, сходный с плацебо. Наиболее частыми побочными явлениями при длительном применении препарата являлись инфекции верхних дыхательных путей, назофарингит, головная боль, диарея. Прибавка массы тела не отмечается. Следует еще раз подчеркнуть, что в большинстве исследований частота развития гипогликемий при лечении иДПП-4 была сходна с плацебо, что объясняется глюкозозависимым действием инкретинов. Гипогликемии главным образом были зафиксированы при совместном применении с ПСМ, что диктует необходимость уменьшения их дозы при комбинации с иДПП-4.
Имелись отдельные сообщения о случаях развития острых панкреатитов, возможно связанных с приемом иДПП-4. Но в большинстве случаев развитие панкреатита ассоциировалось с наличием диабета, ожирения, высокого уровня холестерина и триглицеридов.
ФОРМА ВЫПУСКА И РЕЖИМ ДОЗИРОВАНИЯ Ситаглиптин
Ситаглиптин является высокоселективным иДПП-4 и был первым одобренным к применению препаратом из группы иДПП-4.
Препарат выпускается в таблетированной форме и зарегистрирован в дозах 25, 50 и 100 мг. Обычно рекомендуется прием препарата в дозе 100 мг/сут однократно внутрь, независимо от времени суток и приема пищи. При почечной недостаточности доза ситаглиптина может быть снижена вплоть до 25 мг 1 раз в сутки.
Имеются фиксированные комбинации ситаглиптина с метформином. Сочетание метформина и ситаглиптина позволяет предполагать воздействие на все основные патогенетические механизмы СД 2-го типа: инсулинорезистентность, повышенную продукцию глюкозы печенью и нарушенный ответ β-клеток поджелудочной железы.
В настоящее время в Российской Федерации зарегистрированы комбинированные препараты, содержащие 50 мг ситаглиптина и разные дозировки метформина: 500 мг, или 850 мг, или 1000 мг. Этот комбинированный препарат чаще назначается дважды в день. Комбинация ситаглиптина с метформином обеспечивает удобство применения терапии и повышает комплаентность пациентов.
Вилдаглиптин
Вторым препаратом из группы иДПП-4, появившимся на российском рынке, стал вилдаглиптин.
Вилдаглиптин выпускается в таблетированной форме по 50 мг. Рекомендуемая доза препарата составляет 50 мг 1 или 2 раза в сутки. Вилдаглиптин принимают внутрь независимо от приема пищи. Дозу 50 мг/сут следует назначать в один прием утром. Дозу 100 мг/сут следует назначать по 50 мг 2 раза в сутки утром и вечером.
Имеются фиксированные комбинации вилдаглиптина с метформином. В их состав входят вилдаглиптин в дозе 50 мг и метформин в дозах 500 мг, или 850 мг, или 1000 мг. Этот комбинированный препарат чаще назначается дважды в день.
Саксаглиптин
Препарат выпускается в дозах 5 и 2,5 мг. Назначается один раз в день, чаще всего в дозе 5 мг.
Имеются фиксированные комбинации вилдаглиптина с метформином. В их состав входят саксаглиптин в дозе 2,5 или 5 мг и метформин пролонгированного действия в дозах 500 или 1000 мг. Этот комбинированный препарат чаще назначается один раз в день.
Линаглиптин
Препарат выпускается в дозе 5 мг, назначается в этой дозе один раз в день. Таким образом, не требуется титрация дозы, а также коррекция дозы на любой стадии ХБП.
Алоглиптин
Препарат выпускается в дозах 12,5 и 25 мг. Назначается один раз в день чаще всего в дозе 25 мг.
Ингибиторы натрий-глюкозного котранспортера 2-го типа
С 2012 г. в мире и с 2014 г. в России существует новая группа ЛС для лечения СД 2-го типа с таким механизмом действия. В настоящее время в России зарегистрировано три препарата этой группы: дапаглифлозин, эмпаглифлозин и ка наглифлозин.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
У здоровых людей в клубочках почек за сутки фильтруется примерно 180 г глюкозы, которая подвергается практически полной реабсорбции в проксимальных канальцах. 90% глюкозы реабсорбируются в начальной части проксимального канальца (S1-сегмент). Основным натрий-зависимым переносчиком глюкозы, ответственным за ее реабсорбцию в S1-сегменте, является белок - натрий-глюкозный котранспортер 2-го типа, состоящий из 672 аминокислот и содержащий 14 мембраносвязывающих сегментов. Ингибирование натрий-глюкозного котранспортера 2-го типа посредством не зависимого от инсулина механизма приводит к снижению реабсорбции глюкозы в проксимальных канальцах и увеличению ее экскреции с мочой с последующим снижением уровня глюкозы крови. Второй эффект - это снижение массы тела за счет потери достаточно большого количества энергетического субстрата, которым является глюкоза. Таким образом, эффект эмпаглифлозина не зависит от функционального состояния β-клеток поджелудочной железы и метаболизма инсулина.
ФАРМАКОКИНЕТИКА
Дапаглифлозин обладает высокой (75%) биодоступностью при введении внутрь, быстро всасывается из ЖКТ, имеет линейную фармакокинетику и достаточно длительный период полувыведения (13,8±9,4 ч), позволяющий применять его один раз в сутки.
Эмпаглифлозин после приема внутрь быстро всасывался, максимальная концентрация эмпаглифлозина в плазме крови достигалась через 1,5 ч. Затем концентрация эмпаглифлозина в плазме снижалась в две фазы. Прием пищи не оказывает клинически значимого влияния на фармакокинетику эмпаглифлозина. Объем распределения в период устойчивой концентрации в плазме крови составлял примерно 73,8 л. После перорального применения здоровыми добровольцами меченого эмпаглифлозина [14 C] связывание с белками плазмы составляло 86%. Основной путь метаболизма эмпаглифлозина у человека - глюкуронидация с участием уридин-5'-дифосфоглюкуронозилтрансфераз UGT2B7, UGT1A3, UGT1A8 и UGT1A9. Системное влияние каждого метаболита невелико (менее 10% от общего влияния эмпаглифлозина). Период полувыведения составлял примерно 12,4 ч. В случае применения эмпаглифлозина один раз в день устойчивая концентрация в плазме крови достигалась после пятой дозы. После перорального применения меченого эмпаглифлозина [14 C] у здоровых добровольцев выводилось примерно 96% дозы (через кишечник 41% и почками 54%). Через кишечник большая часть меченого препарата выводилась в неизмененном виде. Почками в неизмененном виде выводилась только половина меченого препарата.
Канаглифлозин имеет сходные фармакокинетические свойства (биодоступность при приеме внутрь - 65%, период полувыведения - 10,6-13,1 ч, высокая степень связывания с белками плазмы, отсутствие выраженного метаболизма цитохромами P450 в печени).
ПОКАЗАНИЯ
СД 2-го типа в качестве монотерапии у пациентов с неадекватным гликемическим контролем только на фоне диеты и физических упражнений, назначение метформина которым считается нецелесообразным ввиду непереносимости; в качестве комбинированной терапии с другими гипогликемическими средствами, включая инсулин, когда применяемая терапия совместно с диетой и физическими упражнениями не обеспечивает необходимого гликемического контроля.
ПРОТИВОПОКАЗАНИЯ
Повышенная чувствительность к любому компоненту препарата, СД 1-го типа, ДКА, редкие наследственные нарушения (дефицит лактазы, непереносимость лактозы, глюкозо-галактозная мальабсорбция), почечная недостаточность (дапаглифлозин при СКФ <60 мл/мин/1,73 м2 ), эмпаглифлозин и канглифлозинρ при СКФ <45 мл/мин/1,73 м2 (в связи с неэффективностью), беременность и период грудного вскармливания, возраст до 18 лет (в связи с недостаточностью данных по эффективности), возраст старше 85 лет, применение в комбинации с аГПП-1 (в связи с отсутствием данных по эффективности и безопасности).
С осторожностью следует применять ингибиторы натрий-глюкозного котранспортера 2-го типа у пациентов с риском развития гиповолемии (применение гипотензивных препаратов со случаями артериальной гипотензии в анамнезе), при заболеваниях ЖКТ, приводящих к потере жидкости, в возрасте старше 75 лет, при инфекциях мочеполовой системы.
ПОБОЧНЫЕ ЭФФЕКТЫ
Наиболее часто встречаемыми побочными эффектами ингибиторов натрий-глюкозного котранспортера 2-го типа являются инфекции мочевыводящих путей и грибковое поражение гениталий (вагинальный кандидоз, вульвовагинит, баланопостит), а также гиповолемия (которая выражалась снижением АД, ортостатической артериальной гипотензией, дегидратацией, обмороком), учащенное мочеиспускание. Частота развития гипогликемий при лечении ингибиторами натрий-глюкозного котранспортера 2-го типа в качестве монотерапии была сходна с плацебо, что объясняется инсулиннезависимым механизмом действия. Гипогликемии главным образом были зафиксированы при совместном применении с ПСМ и инсулином.
ФОРМА ВЫПУСКА И РЕЖИМ ДОЗИРОВАНИЯ Дапаглифлозин
Препарат выпускается в таблетках в дозе 10 мг. Рекомендуемая доза препарата составляет 10 мг 1 раз в сутки. При нарушениях функции печени легкой или средней степени тяжести нет необходимости корректировать дозу препарата. Пациентам с нарушением функции печени тяжелой степени рекомендуется начальная доза препарата 5 мг. При хорошей переносимости доза может быть увеличена до 10 мг.
Эмпаглифлозин
Препарат выпускается в таблетках в дозах 10 и 25 мг. Рекомендуемая начальная доза составляет 10 мг 1 раз в день, внутрь. В случае если суточная доза 10 мг не обеспечивает адекватного гликемического контроля, доза может быть увеличена до 25 мг. Максимальная суточная доза составляет 25 мг. Препарат может приниматься независимо от приема пищи в любое время дня. При пропуске дозы пациенту следует принять препарат, как только он об этом вспомнит. Не следует принимать двойную дозу в один день.
Канаглифлозин
Препарат выпускается в таблетках в дозах 100 и 300 мг.
ВЗАИМОДЕЙСТВИЯ
Дапаглифлозин может усиливать диуретический эффект тиазидных и «петлевых» диуретиков и повышать риск развития обезвоживания и артериальной гипотензии.
Метаболизм дапаглифлозина в основном осуществляется посредством глюкуронидной конъюгации под действием UGT1A9. Дапаглифлозин не ингибиует изоферменты системы цитохрома P450CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4 и не индуцирует изоферменты CYP1A2, CYP2B6 или CYP3A4. В связи с этим не ожидается влияния дапаглифлозина на метаболический клиренс сопутствующих препаратов, которые метаболизируются под действием этих изоферментов. После совместного применения дапаглифлозина и рифампицина, индуктора различных активных транспортеров и ферментов, метаболизирующих ЛС, отмечено снижение системной экспозиции дапаглифлозина на 22%, при отсутствии клинически значимого влияния на суточное выведение глюкозы почками. Клинически значимого влияния при применении с другими индукторами (например, карбамазепином, фенитоином, фенобарбиталом) не ожидается.
После совместного применения дапаглифлозина и мефенамовой кислоты (ингибитора UGT1A9) отмечено увеличение на 55% системной экспозиции дапаглифлозина, но без клинически значимого влияния на суточное выведение глюкозы почками. Дапаглифлозин не влиял на фармакокинетику метформина, пиоглитазона, ситаглиптина, глимепирида, гидрохлоротиазида, буметанида, валсартана, дигоксина (субстрат гликопротеина P), или варфарина (S-варфарин, субстрат изофермента CYP2C9), или на антикоагуляционный эффект, оцениваемый по международному нормализованному отношению. Применение однократной дозы дапаглифлозина 20 мг и симвастатина (субстрата изофермента CYP3A4) приводило к повышению на 19% системной экспозиции симвастатина и на 31% системной экспозиции симвастатиновой кислоты, но это повышение не считается клинически значимым.
Эмпаглифлозин не ингибирует, не инактивирует и не индуцирует изоферменты CYP450. Эмпаглифлозин не ингибирует UGT1A1. Лекарственные взаимодействия эмпаглифлозина и ЛС, являющихся субстратами изоферментов CYP450 и UGT1A1, считаются маловероятными.
Эмпаглифлозин является субстратом для гликопротеина P и белка, определяющего резистентность рака молочной железы (BCRP), но в терапевтических дозах не ингибирует эти белки. На основании данных, полученных в исследованиях in vitro, считается, что способность эмпаглифлозина вступать во взаимодействия с препаратами, которые являются субстратами для гликопротеина P, маловероятна. Эмпаглифлозин является субстратом для органических анионных переносчиков: OAT3, OATP1B1 и OATP1B3, но не является субстратом для органических анионных переносчиков - 1 (OAT1) и органических катионных переносчиков-2 (OCT2). Однако лекарственные взаимодействия эмпаглифлозина с препаратами, являющимися субстратами для вышеописанных белков-переносчиков, считаются маловероятными.
Фармакокинетика эмпаглифлозина не изменяется в случае его совместного применения с метформином, глимепиридом, пиоглитазоном, ситаглиптином, линаглиптином, варфарином, верапамилом, рамиприлом, симвастатином, торасемидом и гидрохлоротиазидом. При совместном применении эмпаглифлозина с гемфиброзилом, рифампицином и пробенецидом отмечалось увеличение значения системной экспозиции эмпаглифлозина на 59, 35 и 53% соответственно, однако данные изменения не считались клинически значимыми.
Аналоги амилина
В России не зарегистрированы.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Прамлинтидρ - синтетический аналог человеческого амилина (островковый амилоидный полипептид). Он замедляет опорожнение желудка, подавляет секрецию глюкагона и снижает аппетит.
ФАРМАКОКИНЕТИКА
Прамлинтидρ при подкожном введении быстро всасывается. Максимальную концентрацию в плазме крови препарат достигает примерно через 20 мин, период полувыведения прамлинтидаρ 48 мин, длительность действия примерно 150 мин. Прамлинтидρ выводится почками. Для введения рекомендуют использовать область живота и бедер, меньше подходит область плеча.
ПОКАЗАНИЯ
Прамлинтидρ показан в качестве дополнительной терапии больным СД 1-го и 2-го типа с постпрандиальной гипергликемией и/или увеличением массы тела на фоне инсулинотерапии.
При СД 1-го типа стартовая доза 15 мкг подкожно 3 раза в сутки непосредственно перед основными приемами пищи (т.е. перед приемом пищи, содержащей минимум 250 ккал или 30 г углеводов), дозу увеличивают на 15 мкг каждые 3 суток до 30-60 мкг.
При СД 2-го типа стартовая доза 60 мкг подкожно 3 раза в сутки непосредственно перед основными приемами пищи, через 3-7 сут при хорошей переносимости (т.е. при отсутствии тошноты) дозу увеличивают до 120 мкг 3 раза в сутки.
После назначения прамлинтидаρ во избежание гипогликемии дозу ИКД или ИУКД, как правило, снижают на 50%. Прамлинтидρ следует вводить с помощью отдельного шприца, его не рекомендуют смешивать с инсулином.
ПРОТИВОПОКАЗАНИЯ
Прамлинтидρ противопоказан при наличии к нему гиперчувствительности, установленном гастропарезе, нарушении распознавания гипогликемии.
ПОБОЧНЫЕ ЭФФЕКТЫ
К характерным побочным эффектам прамлинтидаρ относят гипогликемию и реакции со стороны ЖКТ (тошноту, рвоту, потерю аппетита). Также у некоторых пациентов возникают головная боль, усталость, головокружение, фарингит, боли в суставах, кашель, реакции на месте инъекции.
ВЗАИМОДЕЙСТВИЯ
Прамлинтидρ не следует назначать совместно с ингибиторами α-глюкозидаз и препаратами, влияющими на моторику ЖКТ.
СПИСОК ЛИТЕРАТУРЫ
-
Дедов И.И., Кураева Т.Л., Петеркова В.А. Сахарный диабет у детей и подростков. - М.: ГЭОТАР-Медиа, 2007. - 160 с.
-
Дедов И.И., Петеркова В.А., Кураева Т.Л. Российский консенсус по терапии сахарного диабета у детей и подростков // Сахарный диабет. - 2010. -№ 5. - С. 1-8.
-
Дедов И.И., Шестакова М.В. Алгоритмы специализированной медицинской помощи больным сахарным диабетом. - Вып. 7. - М., 2015. - 112 с.
-
Дедов И.И., Шестакова М.В., Аметов А.С. и др. Консенсус совета экспертов Российской ассоциации эндокринологов по инициации и интенсификации сахаро-снижающей терапии у больных сахарным диабетом 2-го типа // Сахарный диабет. - 2011. - № 4. - С. 6-17.
-
Дедов И.И., Шестакова М.В., Аметов А.С. и др. Инициация и интенсификация сахароснижающей терапии у больных сахарным диабетом 2-го типа: обновление консенсуса совета экспертов Российской ассоциации эндокринологов (2015 г.) // Сахарный диабет. - 2015. - № 1. - С. 4-22.
-
Дедов И.И., Шестакова М.В. Оптимизация и интенсификация инсулинотерапии при СД 2-го типа (клинические рекомендации) // Сахарный диабет. - 2010. - № 5. - С. 9-16.
-
Сахарный диабет: диагностика, лечение, профилактика / Под ред. И.И. Дедова, М.В. Шестаковой. - М.: Медицинское информационное агентство, 2011.
-
Смирнова О.М., Никонова Т.В. Лечение сахарного диабета 1-го типа / Под ред. И.И. Дедова. - М., 2003. - 75 с.
-
AACE comprehensive diabetes management algorithm // Endocrine practice. - 2013. - Vol. 19. - N 2. - P. 327-336.
-
American Diabetes Association. Standards of medical care in diabetes - 2015 // Diabetes Care. - 2015. - Vol. 38 (suppl. 1). - P. 1-93.
-
Buse J.B., Polonsky K.S., Burant C.F. Type 2 Diabetes Mellitus. // Williams Textbook of Endocrinology / H.M. Kronenberg, S. Melmed, K.S. Polonsky, P.R. Larsen (Eds). - Saunders, 2008. - 11th ed. - 1936 p.
-
Davis S.N. Insulin, oral hypoglycemic agents, and the pharmacology of the endocrine pancreas. - 11th ed. - Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. - McGraw-Hill Professional, 2005. - 1984 p.
-
DeWitt D. E., Hirsch I.B. Outpatient insulin therapy in type 1 and type 2 diabetes mellitus: scientific review // JAMA. - 2003. - Vol. 289. - P. 2254-2264.
-
Eisenbarth G.S., Polonsky K.S., Buse J.B. Type 1 Diabetes Mellitus // Williams Textbook of Endocrinology / H.M. Kronenberg, S. Melmed, K.S. Polonsky, P.R. Larsen (Eds). - Saunders, 2008. - 11th ed. - 1936 p.
-
Gardner D.G., Shoback D.M. Greenspan’s Basic and Clinical Endocrinology. - 8th ed. - McGraw-Hill Medical, 2007. - 960 p.
-
International Diabetes Federation. Global Guideline for Type 2 diabetes. - Brussels: International Diabetes Federation, 2012.
-
Inzucchi S.E., Bergenstal R.M., Buse J.B. et al. Management of Hyperglycemia in Type 2 Diabetes, 2015: A Patient-Centered Approach Update to a Position Statement of the American Diabetes Association and the European Association for the Study of Diabetes // Diabetes Care. - 2015. - Vol. 38. - P. 140-149. doi: 10.2337/dc14-2441.
-
Inzucchi S.E., Bergenstal R.M., Buse J.B. et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) // Diabetes Care. - 2012. - Vol. 35 (6). - P. 1364-79.
-
ISPAD Clinical Practice Consensus Guidelines 2014 Compendium // Pediatric Diabetes. - 2014. - Vol. 15 (suppl. 20). - P. 1-290.
-
Kahn C.R., Weir G.C., King G.L. et al. Joslin?s Diabetes Mellitus. - 14th ed. - Lippincott Williams & Wilkins, 2004. - 1224 p.
-
Katzung B.G. Basic and Clinical Pharmacology. - 10th ed. - McGraw-Hill Medical, 2006. - 1179 p.
-
Mooradian A.D., Bernbaum M., Albert S.G. Narrative review: a rational approach to starting insulin therapy // Ann. Intern. Med. - 2006. - Vol. 145. - P. 125-134.
-
Nathan D.M., Davidson M.B., DeFronzo R. A. et al. Impaired Fasting Glucose and Impaired Glucose Tolerance: Implications for care // Diabetes Care. - 2007. - Vol. 30. - P. 753-759.
-
Nesto R.W., Bell D., Bonow R.O., Fonseca V. et al. Thiazolidinedione Use, Fluid Retention, and Congestive Heart Failure // A Consensus Statement From the American Heart Association and American Diabetes Association. Circulation. - 2003. - Vol. 108. - P. 2941-2948.
-
Salpeter S.R., Greyber E., Pasternak G.A. et al. Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitus: systematic review and meta-analysis // Arch. Intern. Med. - 2003. - Vol. 163. - P. 2594-2602.
ПРЕПАРАТЫ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ЩИТОВИДНОЙ ЖЕЛЕЗЫ
Мануйлова Ю.А., Колода Д.Е.
Антитиреоидные средства
Антитиреоидные средства, или тиреостатики, - группа ЛС, которые применяются для угнетения синтеза тиреоидных гормонов (Т4 и Т3 ) при синдроме эндогенного тиреотоксикоза (прежде всего для лечения диффузного токсического зоба). В разных странах частота использования тиреостатиков неодинакова. Например, в США предпочитают РЙТ, закономерный ее итог - развитие постлучевого гипотиреоза, который впоследствии без особых усилий корректируется назначением заместительной терапии препаратами гормонов щитовидной железы (обычно левотироксина натрия). В России, как и в Европе, традиционно применяют тиреостатические ЛС из группы тионамидов, так как имеющийся в этих странах легкий дефицит йода повышает чувствительность к тиреостатикам. Другие антитиреоидные препараты (калия перхлорат, лития карбонат, калия йодид, натрия йодид) в настоящее время во всем мире для лечения тиреотоксикоза используют редко.
КЛАССИФИКАЦИЯ
По механизму действия выделяют четыре группы антитиреоидных средств:
-
нарушающие транспорт йода внутрь фолликулов (калия перхлорат);
-
нарушающие синтез тиреоидных гормонов (тионамиды: тиамазол, карбимазолρ , пропилтиоурацил);
-
ингибирующие высвобождение тиреоидных гормонов (йодиды в фармакологических дозах, лития карбонат);
-
разрушающие фолликулы щитовидной железы (радиоактивный йод). Тионамиды (производные тиомочевины) содержат тионамидную группу.
В настоящее время применяют только два препарата на основе имидазольного кольца - тиамазол [1] и карбимазолρ , а также пропилтиоурацил, имеющий пиримидиновое кольцо. В организме человека карбимазолρ полностью превращается в тиамазол.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Тионамиды обладают общей способностью блокировать тиреоидную пероксидазу - ключевой фермент синтеза тиреоидных гормонов. Тиреоидная пероксидаза участвует во всех трех этапах синтеза: окислении поступающего в фолликул йода с образованием активных промежуточных соединений, йодировании остатков тирозина в молекуле тиреоглобулина и соединении молекул йодотирозинов внутри тиреоглобулина с образованием Т4 и Т3 . Под действием антитиреоидных ЛС деятельность тиреоидной пероксидазы нарушается, в результате снижается концентрация гормонов щитовидной железы в периферической крови. Из описания механизма действия становится понятно, что тиреостатики не способны инактивировать уже существующий в организме избыток тиреоидных гормонов, а также неэффективны при экзогенном тиреотоксикозе.
Помимо тиреостатического действия, эти ЛС оказывают иммуномодулирующее влияние на ткань щитовидной железы. При этом снижается экспрессия тиреоидных антигенов, уменьшается высвобождение простагландинов и цитокинов. Кроме того, тионамиды тормозят образование свободных радикалов кислорода в Т- и В-лимфоцитах и особенно в антиген-представляющих клетках. Тиамазол стимулирует экспрессию Fas-лиганда, тем самым запуская апоптоз Т-лимфоцитов, инфильтрирующих ткань щитовидной железы. В итоге под действием тиреостатиков в периферической крови снижается концентрация антитиреоидных антител, а в ткани щитовидной железы уменьшается лимфоидная инфильтрация.
Пропилтиоурацил тормозит периферическую конверсию Т4 в биологически более активный Т3 за счет ингибирования дейодиназы 1-го типа в периферических тканях. В связи с этим большие дозы пропилтиоурацила позволяют быстро нормализовать состояние больного при тяжелом тиреотоксикозе (рис. 4.5, табл. 4.14).
ФАРМАКОКИНЕТИКА
Тионамиды хорошо и быстро всасываются из ЖКТ. Терапевтическая концентрация пропилтиоурацила в крови достигается уже через 20-30 мин после приема внутрь. Карбимазолρ и тиамазол обладают почти одинаковыми фармакокинетическими свойствами. При попадании в щитовидную железу карбимазолρ полностью превращается в тиамазол за счет отщепления пропильной группы (15 мг карбимазолаρ соответствуют 10 мг тиамазола). Тиреостатики накапливаются в ткани щитовидной железы, где со временем их концентрация становится значительно выше, чем в плазме, в связи с этим их действие сохраняется некоторое время и после отмены. Тионамиды в основном метаболизируются в печени, где подвергаются глюкуронизации. Проникновение через плаценту и в грудное молоко в большей степени характерно для тиамазола, поэтому в I триместре беременности предпочтительнее назначение пропилтиоурацила, однако из-за его гепатотоксичности во II и III триместрах беременности рекомендован тиамазол (пропилтиоурацил во время лактации является препаратом второй линии) (табл. 4.15).
ПОКАЗАНИЯ
Антитиреоидные ЛС занимают одно из ведущих мест в лечении тиреотоксикоза различной этиологии, причем и тиамазол, и пропилтиоурацил применяются одинаково успешно, однако в настоящее время основным показанием для применения пропилтиоурацила остается I триместр беремености. Различия между ними только в относительной эффективной дозе: у тиамазола она в 10 раз меньше, чем у пропилтиоурацила.
У пациентов с диффузным токсическим зобом, которым впервые поставлен этот диагноз, при отсутствии показаний к оперативному лечению или РЙТ, консервативное лечение тиреостатиками - метод выбора (см. «Синдром тиреотоксикоза»).
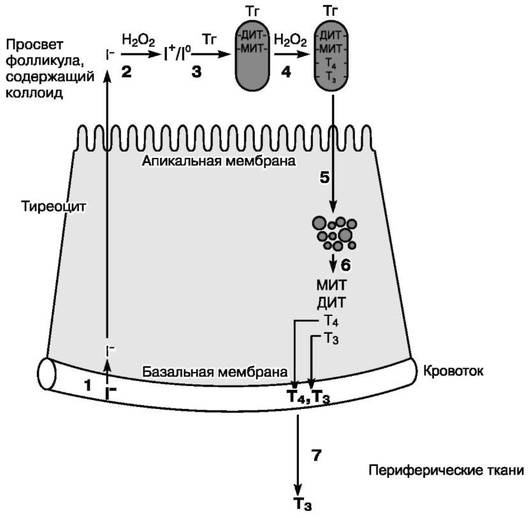
| Этап | Основной фермент | Ингибиторы |
|---|---|---|
1. Транспорт йодидов из внеклеточного пространства внутрь тиреоцита |
Na+-, I--симпортер, связанный с Na+-, K+- зависимой АТФазой |
Перхлорат (ClO4-), тиоцианат (SCN-) |
2. Окисление йодидов с образованием активных промежуточных соединений |
Тиреоидная пероксидаза |
Тионамиды |
3. Йодирование остатков тирозина в молекуле тиреоглобулина |
Тиреоидная пероксидаза |
Тионамиды, большие дозы йодидов |
4. Конденсация (соединение) йодотирозинов внутри тиреоглобулина с образованием Т4 и Т3 |
Тиреоидная пероксидаза |
Тионамиды |
5. Эндоцитоз коллоида |
Аденилатциклаза, связанная с рецептором ТТГ |
Литий, большие дозы йодидов |
6. Протеолиз тиреоглобулина |
Протеазы |
Большие дозы йодидов |
7. Дейодирование Т4 |
5'-дейодиназа |
Пропилтиоурацил, натрия йоподат, β-адреноблокаторы, глюкокортикоиды, амиодарон |
| ЛС | Биодоступность, % | Связывание с белками плазмы, % | Период полувыведения, ч | Продолжительность действия, ч | Путь элиминации | Трансплацентарный транспорт | Отношение концентраций грудное молоко/ плазма крови |
|---|---|---|---|---|---|---|---|
Тиaмaзoл |
80-95 |
0 |
4-6 |
24 и более |
Печеночный |
Низкий |
1 |
Прoпилтиoурaцил |
80-95 |
75-80 |
1-2 |
12-24 |
Печеночный |
Очень низкий |
0,1 |
В литературе обсуждается вопрос о стартовой дозе тиреостатиков. Общепринято назначать относительно высокие дозы - 30-40 мг тиамазола. Некоторые тиреои-дологи рекомендуют вместо этого использовать более низкие дозы (для предотвращения побочных эффектов) - 15-20 мг тиамазола. Успешность использования режима малых доз подтверждена результатами Европейского многоцентрового исследования тиреостатической терапии (European Multicenter Study Group on Antithyroid Drug Treatment), завершенного в 1993 г.
В качестве поддерживающего лечения используют два режима: низкие дозы тиреостатиков (схема «блокируй») либо более высокие дозы в сочетании с приемом левотироксина натрия (схема «блокируй и замещай»). Критерием эффективности лечения является нормализация уровня свободного Т4 , что происходит в среднем через 3-4 нед лечения, при этом уровень ТТГ еще длительное время может оставаться подавленным (до 6 мес).
При назначении тионамидов тиреостатический эффект развивается только спустя несколько недель, поэтому на первое время параллельно назначаются β-адреноблокаторы, которые быстро купируют тахикардию и вегетативную симптоматику. Примерно через 2-4 нед дозу β-адреноблокаторов начинают постепенно снижать, иногда до полной отмены.
Длительность лечения тиреостатиками должна быть не более 12-18 мес, поскольку дальнейший их прием не оказывает существенного влияния на прогноз заболевания. При наличии противопоказаний или развитии непереносимости тионамидов, а также в случае рецидива заболевания после проведенной тиреостатической терапии следует рассмотреть вопрос о радикальном лечении: оперативном или радиоактивным йодом. Однако перед операцией рекомендуется достичь эутиреоза с помощью тиреостатических ЛС.
У больных с декомпенсированной функциональной автономией щитовидной железы тиреостатики показаны только в качестве подготовки к операции.
Тиреостатики могут использоваться для паллиативного лечения эндогенного тиреотоксикоза (диффузного токсического зоба, функциональной автономии), если пациент по тем или иным причинам отказывается от операции или РЙТ.
При развитии у пациента тиреотоксического криза лечение проводят большими дозами тиреостатиков, причем таблетированные препараты можно измельчить и вводить через назогастральный зонд, так как в настоящее время нет ЛС из группы тионамидов для внутривенного введения. Предпочтение отдается пропилтио-урацилу в связи с его дополнительной способностью блокировать периферическую конверсию Т4 в Т3 .
ПРОТИВОПОКАЗАНИЯ
Противопоказанием к назначению тиреостатиков из группы тионамидов является повышенная чувствительность к ним. Следует подчеркнуть, что беременность и грудное вскармливание не являются абсолютным противопоказанием к использованию тионамидов. Тионамиды по риску применения во время беременности относятся к классу D, поэтому у беременных и кормящих женщин они могут использоваться под строгим наблюдением врача в качестве альтернативы оперативному лечению. Однако беременность является абсолютным противопоказанием для использования схемы «блокируй и замещай», так как левотироксин натрия не проникает, а тиреостатики легко проходят через плаценту и могут вызвать тяжелый гипотиреоз у плода. Перед назначением антитиреоидных ЛС беременным необходимо провести дифференциальную диагностику между диффузным токсическим зобом и транзиторным тиреотоксикозом беременных. Последний возникает в ответ на стимуляцию щитовидной железы ХГЧ и не требует лечения.
ПОБОЧНЫЕ ЭФФЕКТЫ
Наиболее грозным осложнением применения тионамидов является агранулоцитоз. В настоящее время не рекомендуется постоянное мониторирование уровня лейкоцитов, а предлагается лишь обязательно информировать больного о том, что при повышении температуры, появлении болей в горле или диспепсических явлений он должен обратиться к врачу. В этой ситуации следует срочно определить количество лейкоцитов, при показателе менее 3×109 /л лечение тиреостатиками должно быть прервано, необходимо назначить стимуляторы лейкопоэза (ленограстим, филграстим). Наиболее часто агранулоцитоз развивается у лиц старше 40 лет, принимающих более 40 мг тиамазола в сутки.
Применение тионамидов сопряжено с возможностью развития тромбоцитопении и повышения риска спонтанных кровотечений, поэтому необходимо контролировать протромбиновое время, особенно перед хирургическими манипуляциями.
Возникающие в процессе лечения кожные высыпания различного генеза не следует автоматически считать аллергической реакцией на ЛС. Ее можно подтвердить лишь с помощью кожной пробы.
При лечении высокими дозами тиреостатиков без заместительной терапии левотироксином натрия на фоне развития гипотиреоза может сформироваться зоб («зобогенный» эффект тиреостатиков) за счет гиперстимуляции ТТГ щитовидной железы.
Как правило, при использовании малых доз ЛС опасные осложнения почти не встречаются, низка и общая частота всех осложнений. Побочные эффекты тионамидов
При передозировке могут возникать тошнота, рвота, головная боль, лихорадка, артралгии, панцитопения, поражение печени, нейропатии, угнетение или возбуждение. В качестве лечения проводят промывание желудка, назначают активированный уголь и симптоматическую терапию.
ВЗАИМОДЕЙСТВИЯ
Тионамиды способны вступать в фармакодинамические и фармакокинетические взаимодействия со многими ЛС. Они могут усиливать действие антикоагулянтов, повышают риск развития побочных эффектов β-адреноблокаторов, сердечных гликозидов, нарушают выведение теофиллина.
Следует по возможности избегать одновременного назначения с ними ЛС, способных вызывать агранулоцитоз (НПВС, клозапин, сульфасалазин и др.).
СПИСОК ЛИТЕРАТУРЫ
-
Фадеев В.В. Заболевания щитовидной железы в регионе легкого йодного дефицита. - М.: Издательский дом «Видар-М», 2005. - 240 с.
-
Basic and Clinical Pharmacology / B.G. Katzung (Ed.). - McGraw-Hill Medical, 2006. - 10th ed. - 1179 p.
-
Braverman L.E. Diseases of the thyroid. - Humana Press, 2002. - 2nd ed. - 400 p.
-
Davies T.F., Larsen P.R. Thyrotoxicosis // Williams Textbook of Endocrinology / H.M. Kronenberg, S. Melmed, K.S. Polonsky, P.R. Larsen (Eds). - Saunders, 2008. - 11th ed. - Р. 333-376.
-
De Groot L., Abalovich M., Alexander E.K. et al. Management of thyroid dysfunction during pregnancy and postpartum: an Endocrine Society clinical practice guideline // J. Clin. Endocrinol. Metab. - 2012. - Vol. 97 (8). - P. 2543-2565.
-
Farwell A.P., Braverman L.E. Thyroid And Antithyroid Drugs / L. Brunton, J. Lazo, K. Parker (Eds). Goodman & Gilman?s The Pharmacologic Basis of Therapeutics. - McGraw- Hill Professional, 2005. - 11th ed. - 1984 p.
-
Gartner R., Haen E. Endokrinpharmakologie. Pharmakotherapie mit Hormonen / W. Forth, Еd. Allgemeine und spezielle Pharmakologie und Toxikologie, 8. Aufl. - München: Urban & Fischer, 2001. - P. 671-737.
-
Greenspan?s Basic and Clinical Endocrinology / D.G. Gardner, D.M. Shoback (Eds). - McGraw-Hill Medical, 2007. - 8th ed. - 960 p.
-
Reinwein D., Benker G., Lazarus J.H., Alexander W.D. European Multicenter Study Group on Antithyroid Drug Treatment. A prospective randomized trial of antithyroid drug dose in Graves? disease therapy // J. Clin. Endocrinol. Metab. - 1993. - Vol. 76. - P. 1516-1521.
Препараты гормонов щитовидной железы
Впервые Т4 из гидролизата щитовидной железы выделил Э.К. Кендалл в 1915 г. Спустя 11 лет Харрингтон установил химическую структуру Т4 , а в 1927 г. совместно с Баргером впервые синтезировал этот гормон. Т3 был получен Гроссом и Питт-Риверсом лишь в 1952 г.
В клинической практике в качестве источника тиреоидных гормонов длительное время использовали препараты щитовидной железы различных животных (обычно крупного рогатого скота). Однако подобные ЛС было трудно стандартизировать, поэтому в настоящее время повсеместно используют тиреоидные гормоны, полученные синтетическим путем.
КЛАССИФИКАЦИЯ
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Тиреоидные гормоны действуют практически во всех тканях организма. Основные эффекты этих гормонов - регуляция энергетического, белкового, углеводного и жирового обмена, роста и развития организма в целом, влияние на сердечно-сосудистую, костно-мышечную системы и ЦНС.
Только Т3 (лиотиронин) - истинный гормон, только он способен оказывать действие на клеточном уровне. А Т4 [левотироксин натрия (L-тироксин♠ )] лишь прогормон, в периферических тканях он трансформируется в Т3 . Клетки способны самостоятельно регулировать процесс конверсии Т4 в Т3 , определяя количество активного гормона в своем окружении. Превращение Т4 в Т3 происходит за счет дейодирования наружного тирозильного кольца Т4 под действием особых ферментов дейодиназ.
Тиреоидные гормоны проникают внутрь клеток путем диффузии. Внутри клеток происходит связывание Т3 со специфическими ядерными рецепторами для тиреоидных гормонов (существуют подтипы α и β). Образующийся комплекс «гормон - рецептор» воздействует на определенные регуляторные участки дезок-сирибонуклеиновой кислоты (ДНК). Данный процесс невозможен без участия особых белков-кофакторов. В итоге увеличивается транскрипционная активность РНК, активируется синтез белка и повышается внутриклеточный транспорт глюкозы и аминокислот. Как следствие, увеличивается потребление кислорода почти во всех тканях организма, повышается основной обмен и усиливается теплопродукция (калоригенный эффект).
Повышение внутриклеточного транспорта глюкозы происходит за счет потенцирования действия инсулина. В то же время тиреоидные гормоны стимулируют расщепление инсулина, в связи с чем при тиреотоксикозе развивается постпрандиальная гипергликемия.
Тиреоидные гормоны оказывают положительное хроно- и инотропное действие на миокард, увеличивают количество катехоламиновых рецепторов в миокарде и повышают их чувствительность, увеличивают потребность миокарда в кислороде. Именно поэтому избыток тиреоидных гормонов может вызывать тахикардию, мерцательную аритмию и правожелудочковую недостаточность.
Избыток тиреоидных гормонов стимулирует преимущественно резорбтивные процессы в костной системе.
Кроме того, под влиянием тиреоидных гормонов активируются процессы глюконеогенеза и гликогенолиза, стимулируется эритропоэз, ускоряется метаболизм и выведение других гормонов и различных ЛС, активизируется деятельность дыхательного центра.
В процессе внутриутробного развития плода, а также в постнатальном периоде тиреоидные гормоны играют важнейшую роль в правильном формировании ЦНС и опорно-двигательного аппарата. Недостаток этих гормонов приводит к серьезной задержке психомоторного и физического развития ребенка.
Тиреоидные гормоны угнетают синтез и секрецию ТТГ гипофизом за счет механизма «отрицательной обратной связи». Уже при минимальном снижении уровня Т4 , которое даже не выходит за пределы нормальных значений, определяемых лабораторными методами, происходит повышение содержания ТТГ. Предполагается, что тиреоидные гормоны также угнетают образование тиреотропин-рилизинг-гормона в гипоталамусе.
ФАРМАКОКИНЕТИКА
Тиреоидные гормоны хорошо всасываются в ЖКТ. Однако следует учитывать, что абсорбция может повышаться при голодании и, наоборот, значительно снижаться при смешивании с пищей. Многие продукты питания, особенно пищевые волокна, препятствуют полноценному всасыванию Т4 и Т3 . В связи с этим дозу ЛС, содержащих эти гормоны, рекомендуют принимать однократно в одно и то же время суток (обычно утром) за 30-40 мин до еды и с интервалом как минимум 4 ч до или после приема некоторых ЛС или витаминов. С возрастом абсорбция Т4 и Т3 снижается.
При поступлении в организм основная масса тиреоидных гормонов сразу связывается с белками-переносчиками, образуя своего рода гормональное «депо». Существуют три основных белка-переносчика тиреоидных гормонов: тироксинсвязывающий глобулин, тироксинсвязывающий преальбумин (или транстиретин) и альбумин. Около 75% количества Т4 связывается с тироксинсвязывающим глобулином, 15-20% - с тироксинсвязывающим преальбумином и 5% - с альбумином. Т3 главным образом связывается с тироксинсвязывающим глобулином. Поскольку Т3 по сравнению с Т4 обладает меньшим сродством к белкам-переносчикам, процент свободной формы у него значительно выше (0,4 и 0,04% соответственно). Поэтому Т3 раньше начинает действовать и меньшее время сохраняется в организме, чем Т4 . Несмотря на то что лишь такая малая доля тиреоидных гормонов является «свободной», именно она играет физиологическую роль, так как только не связанные с белками Т3 и Т4 обладают способностью проникать внутрь клетки и взаимодействовать со специфическими рецепторами.
«Свободные» и «связанные» гормоны находятся в состоянии динамического равновесия. Степень сродства транспортных белков изменяется в зависимости от концентрации «свободных» Т3 и Т4 в крови.
Как было сказано ранее, в периферических тканях происходит конверсия Т4 в Т3 , который активнее в 3-5 раз. В связи с этим монотерапия левотироксином натрия (препаратом Т4 ) способна полностью обеспечить организм необходимым количеством тиреоидных гормонов, на ее фоне концентрация Т3 в плазме крови увеличивается постепенно в течение нескольких недель.
При назначении лиотиронина (препарата Т3 ) в качестве монотерапии или в составе комбинированных средств (левотироксин натрия/лиотиронин) происходит резкий подъем уровня Т3 в крови, который нормализуется лишь спустя 2-4 ч. При таком лечении организм пациента несколько часов в день находится в состоянии медикаментозного тиреотоксикоза, что может приводить к развитию осложнений (например, со стороны сердечно-сосудистой системы).
Период полувыведения тиреоидных гормонов в определенной степени зависит от функционального состояния щитовидной железы: при гипертиреозе он увеличивается, а при гипотиреозе уменьшается. Выводятся тиреоидные гормоны в основном почками.
Некоторые фармакокинетические параметры тиреоидных гормонов представлены в табл. 4.16.
ПОКАЗАНИЯ
Основная сфера применения тиреоидных гормонов - заместительная терапия различных форм гипотиреоза. Метод выбора в этом случае - монотерапия левотироксином натрия (см. «Синдром гипотиреоза»).
| ЛС | Биодоступность, % | Связывание с белками плазмы, % | Период до начала действия, ч | Период полувыведения, сут | Пик действия, сут | Продолжительность действия, сут |
|---|---|---|---|---|---|---|
Левотироксин натрия |
75-85 |
99,96 |
48-72 |
6-7 |
9-10 |
10-15 |
Лиотиронин |
90-100 |
99,6 |
6-12 |
1-2 |
2-3 |
5-10 |
При манифестном гипотиреозе у взрослых расчетная заместительная доза левотироксина натрия составляет 1,6-1,8 мкг на 1 кг фактической массы тела. При субклиническом гипотиреозе эта доза несколько ниже - обычно 0,9-1 мкг/кг.
Цель заместительной терапии первичного гипотиреоза - поддержание уровня ТТГ в пределах нормальных значений. Содержание ТТГ медленно меняется после изменения дозы левотироксина натрия, поэтому его исследуют не ранее чем через 6-8 нед. При адекватной терапии полная нормализация концентрации ТТГ происходит лишь спустя 4-6 мес после начала лечения. Уровень ТТГ незначительно колеблется в течение суток и не изменяется после приема препаратов тиреоидных гормонов утром до забора крови.
Цель заместительной терапии вторичного гипотиреоза - поддержание уровня свободного Т4 в верхней трети интервала нормальных значений. Утром перед забором крови для определения концентрации свободного Т4 не следует принимать левотироксин натрия, иначе содержание свободного Т4 в крови будет значимо повышенным (на 15-20%) примерно в течение 9 ч после приема ЛС.
Молодым пациентам с гипотиреозом и без тяжелых сопутствующих заболеваний можно назначить всю расчетную дозу сразу. С возрастом потребность в тиреоидных гормонах снижается, вероятно, за счет уменьшения клиренса левотироксина натрия, в связи с чем пожилым пациентам может потребоваться снижение дозы.
У пожилых пациентов, имеющих заболевания сердечно-сосудистой системы, лечение левотироксином натрия начинают с дозы 12,5-25 мкг. С интервалом два месяца дозу увеличивают на 12,5-25 мкг до нормализации уровня ТТГ. При появлении или ухудшении симптомов заболеваний сердца необходимо провести коррекцию терапии.
Экзогенные тиреоидные гормоны по риску применения во время беременности относятся к классу А, поэтому их использование у беременных при наличии показаний абсолютно безопасно. Тиреоидные гормоны проникают в грудное молоко в незначительном количестве, поэтому их можно применять во время грудного вскармливания.
При впервые выявленном во время беременности субклиническом или манифестном гипотиреозе следует сразу назначить полную заместительную дозу левотироксина натрия. Беременным с гипотиреозом левотироксин натрия назначают в дозе 2,3 мкг/кг. Если гипотиреоз у пациентки был диагностирован до беременности и она уже получает левотироксин натрия, дозу следует увеличить на 30-50% (обычно на 50 мкг/сут). Строгое выполнение этих рекомендаций особенно важно в первой половине беременности, когда собственная щитовидная железа плода еще не функционирует. Такое лечение позволяет избежать тяжелых психоневрологических нарушений у ребенка.
Потребность в левотироксине натрия у детей значительно выше, чем у взрослых (табл. 4.17). Начальная доза ЛС и время достижения полной заместительной дозы определяются индивидуально в зависимости от возраста, массы тела и наличия сопутствующей патологии сердца.
| Возраст | Доза левотироксина натрия, мкг/кг в сутки |
|---|---|
0-3 мес |
10-15 |
8-10 (для недоношенных) |
|
3-6 мес |
8-10 |
6-12 мес |
6-8 |
1-5 лет |
5-6 |
6-12 лет |
4-5 |
12-18 лет |
2-3 |
Целесообразность использования комбинированных препаратов тиреоидных гормонов (левотироксин натрия/лиотиронин) продолжает обсуждаться.
Переход на комбинированную терапию можно рекомендовать пациентам, у которых, несмотря на компенсацию гипотиреоза на фоне монотерапии левотироксином натрия, сохраняется неудовлетворительное самочувствие и симптомы гипотиреоза. При переводе пациента на комбинированную терапию дозу левотироксина натрия уменьшают на 25-50 мкг и добавляют 10-12,5 мкг лиотиронина. В итоге соотношение левотироксин натрия/лиотиронин должно соответствовать физиологическому (13-20:1), чтобы избежать вышеописанного резкого подъема уровня Т3 в крови после приема ЛС (учитывая соотношение Т4 :Т3 1:13 в имеющихся комбинированных препаратах, целесообразнее использовать отдельные препараты левотироксина натрия и лиотиронина).
Монотерапию гипотиреоза лиотиронином проводят крайне редко.
При гипотиреоидной (микседематозной) коме внутривенно вводят тиреоидные гормоны и глюкокортикоиды. В связи с отсроченным началом действия левотироксина натрия в первые сутки дополнительно вводится лиотиронин внутрь через желудочный зонд (внутривенное введение лиотиронина опасно развитием тяжелых сердечно-сосудистых осложнений; см. «Гипотиреоидная кома»).
В лечении болезни Грейвса-Базедова левотироксин натрия используют в рамках схемы «блокируй и замещай» для компенсации медикаментозного гипотиреоза, развивающегося на фоне длительного приема тиреостатиков (см. «Болезнь Грейвса-Базедова»).
Тиреоидные гормоны (чаще левотироксин натрия) также применяют и для супрессивной терапии, подавления секреции ТТГ до 0,1-0,5 мМЕ/л при различных заболеваниях щитовидной железы. Доза левотироксина натрия, назначаемая после оперативного лечения по поводу высокодифференцированного РЩЖ, составляет 2,2-2,8 мкг/кг массы тела, через 5 лет при отсутствии указаний на рецидив рака пациенты переводятся на заместительную терапию (см. «Рак щитовидной железы»).
ПРОТИВОПОКАЗАНИЯ
Противопоказания к началу терапии тиреоидными гормонами включают декомпенсированный тиреотоксикоз, нелеченую НН и острый инфаркт миокарда. Относительными противопоказаниями считают нарушения ритма сердца, нестабильную стенокардию, миокардиты. Однако при перечисленных состояниях лечение манифестного гипотиреоза может проводиться на фоне надлежащей терапии сердечной патологии.
ПОБОЧНЫЕ ЭФФЕКТЫ
Как правило, при использовании малых и средних доз ЛС тиреоидных гормонов побочные эффекты почти не встречаются. При передозировке или слишком быстром увеличении дозы наблюдаются симптомы тиреотоксикоза (см. «Синдром тиреотоксикоза»).
У больных ИБС тиреоидные гормоны, особенно лиотиронин, могут провоцировать учащение приступов стенокардии, поэтому таким пациентам рекомендуется начинать лечение с малых доз (12,5-25 мкг). Повышение дозы проводится постепенно с интервалом 2-4 нед под контролем электрокардиографии (ЭКГ).
Длительная супрессивная терапия тиреоидными гормонами может приводить к снижению МПК, особенно у женщин в постменопаузе.
ВЗАИМОДЕЙСТВИЯ
Многие ЛС взаимодействуют с тиреоидными гормонами и влияют на функцию щитовидной железы. Однако лишь некоторые взаимодействия имеют значение в клинической практике, когда они либо требуют изменить дозу тиреоидных гормонов, либо влияют на интерпретацию результатов диагностических исследований (табл. 4.18).
| Механизм действия | ЛС | Эффект |
|---|---|---|
Подавление Т4→ Т3 5'-дейодирования |
β-Адреноблокаторы, контрастные средства (иопановая кислота, натриевая соль иоподовой кислоты), амиодарон, глюкокортикоиды |
↓Т3 |
Изменение связывания Т4 и Т3 с белками плазмы |
Эстрогены, героин, метадон, клофибрат, фторурацил (5-фторурацил♠), перфеназин, тамоксифен, ралоксифен, митотан |
↑ тироксинсвязывающего глобулина |
Изменение уровня тироксинсвязывающего глобулина |
Андрогены, глюкокортикоиды, L-аспарагиназа |
↓ тироксинсвязывающего глобулина |
Вытеснение Т4 и Т3 из связи с белками плазмы |
Гепарин натрия, фуросемид, фенитоин, карбамазепин, нПВc, сальсалаты, клофибрат |
↑ свободного Т4 в тест-системе |
Усиление печеночного метаболизма Т4 и Т3 |
Барбитураты, фенитоин, карбамазепин, рифампицин, сертралин |
↓Т4 и Т3 |
Нарушение кишечной абсорбции Т4 |
Алюминия гидрохлорид сукральфат, железа сульфат, колестирамин, колестипол, кальция карбонат, препараты сои, ралоксифен |
↓Т4 и Т3 |
Вытеснение Т4 из тканевого пула |
Циклофосфамид, оральные холецистографические контрастные вещества |
↑Т4 (транзиторно) |
Подавление продукции и секреции ТТГ гипофизом |
Допамин, добутамин, глюкокортикоиды, октреотид, интерферон альфа, бромокриптин, фенитоин |
↓ТТГ |
Под действием эстрогенов и некоторых других ЛС в печени активируется синтез тироксинсвязывающего глобулина, что приводит к увеличению связанной фракции тиреоидных гормонов, соответственно повышаются уровни общих Т4 и Т3 . При этом концентрация свободных тиреоидных гормонов может уменьшаться, что иногда может требовать увеличения принимаемой дозы ЛС. Андрогены, наоборот, уменьшают уровень тироксинсвязывающего глобулина.
У пациентов, получающих гепарин натрия, может быть выявлен неадекватно высокий уровень свободного Т4 в результате образования во время хранения или инкубации образцов сыворотки неэтерифицированных жирных кислот, которые вытесняют Т4 из связи с тироксинсвязывающим глобулином.
Замедление абсорбции Т4 , а также ускорение его разрушения и выведения, вызванное ЛС, принимаемым совместно с ним, может привести к декомпенсации гипотиреоза, поэтому часто требует повышения дозы Т4 .
При длительном лечении фенитоином уровни свободного Т4 и ТТГ могут оказаться похожими на таковые при вторичном гипотиреозе за счет усиления выведения Т4 , с одной стороны, и подавления секреции ТТГ - с другой.
При терапии дофаминомиметиками и глюкокортикоидами уровень ТТГ может оказаться сниженным по отношению к уровню свободного Т4 .
Следует помнить, что развитие «синдрома эутиреоидной патологии» (патологические результаты исследования функции щитовидной железы у пациентов с тяжелыми соматическими заболеваниями при отсутствии реальной тиреоидной патологии) часто связано с эффектами ЛС. Данные о функции щитовидной железы у пациентов в критическом состоянии необходимо трактовать только с учетом подробной информации о назначении таких распространенных ЛС, как дофаминомиметики, глюкокортикоиды, контрастные средства, β-адреноблокаторы, фуросемид и гепарин натрия.
Тиреоидные гормоны сами способны влиять на действие других ЛС (табл. 4.19).
| ЛС | Эффект |
|---|---|
Антикоагулянты (вaрфaрин) |
↑ действия антикоагулянтов |
Антидепрессанты |
↑ действия антидепрессантов |
Сердечные гликозиды (дигоксин) |
↓ действия сердечных гликозидов |
Перорaльные сaхaроснижaющие препараты |
↓ действия перорaльных сaхaроснижaющих препаратов |
СПИСОК ЛИТЕРАТУРЫ
-
Фадеев В.В., Мельниченко Г.А. Гипотиреоз: Руководство для врачей. - М., 2002. - 216 с.
-
Basic and Clinical Pharmacology / B.G. Katzung (Ed.). - McGraw-Hill Medical, 2006. - 10th ed. - 1179 p.
-
Braverman L.E. Diseases of the thyroid. - Humana Press, 2002. - 2nd ed. - 400 p.
-
Farwell A.P., Braverman L.E. Thyroid And Antithyroid Drugs // L. Brunton, J. Lazo, K. Parker (Eds). Goodman & Gilman?s The Pharmacologic Basis of Therapeutics. - McGraw-Hill Professional, 2005. - 11th ed. - 1984 p.
-
Gartner R., Haen E. Endokrinpharmakologie. Pharmakotherapie mit Hormonen // W. Forth, Еd. Allgemeine und spezielle Pharmakologie und Toxikologie, 8. Aufl. - München: Urban & Fischer, 2001. - Р. 671-737.
-
Greenspan?s Basic and Clinical Endocrinology / D.G. Gardner, D.M. Shoback (Eds). - McGraw-Hill Medical, 2007. - 8th ed. - 960 p.
-
Larsen P.R., Davies T.F. Hypothyroidism and Thyroiditis // Williams Textbook of Endocrinology / H.M. Kronenberg, S. Melmed, K.S. Polonsky, P.R. Larsen (Eds). - Saunders, 2008. - 11th ed. - 1936 p.
-
Stockigt J.R. Drug effects on thyroid function // Thyroid international. - 1999. - Р. 5.
-
Wiersinga W.M., Duntas L., Fadeyev V. et al. ETA Guidelines: The Use of L-T4 + L-Т3 in the Treatment of Hypothyroidism // Eur. Thyroid. J. - 2012. - N 1. - P. 55-71.
Препараты йода
КЛАССИФИКАЦИЯ
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Действие йода на синтез тиреоидных гормонов зависит от его дозы. При нарастании поступления йода в организм сначала происходит повышение его органификации (йодирования остатков тирозина в молекуле тиреоглобулина). Однако при достижении определенной критической концентрации высокие дозы йода блокируют собственную органификацию. Следовательно, существует две фазы влияния нарастающих доз йода на синтез гормонов.
В физиологических дозах (до 1000 мкг в день) йод как микроэлемент включается в процесс синтеза тиреоидных гормонов. Помимо этого, йод обладает важнейшим свойством: поступая в достаточном количестве, он предотвращает развитие зоба (увеличения щитовидной железы). Механизм антизобного действия продолжает изучаться, тем не менее на сегодняшний день существует следующая теория. Внутри тиреоцитов, помимо йодтиронинов, формируются соединения йода с липидами (йодлактоны). Йодированные липиды - мощные ингибиторы продукции ИФР-I и других ростовых факторов. При их недостатке факторы роста запускают пролиферативные процессы, приводящие к гиперплазии тиреоцитов. Кроме того, йодированные липиды подавляют в тиреоците цАМФ-зависимые процессы, что, собственно, и предотвращает стимулирующие эффекты ТТГ в условиях достаточного интратиреоидного содержания йода.
Применение высоких, так называемых фармакологических, доз йода (более 0,1 мг/кг массы тела, т.е. более 5 мг в день) обычно в составе таких ЛС, как йод + [калия йодид + глицерол] (Люголя раствор с глицерином♠ ), оказывает противоположное действие, что получило название эффекта Вольфа-Чайкова.
Существует несколько механизмов развития данного феномена. Во-первых, высокая концентрация йода внутри тиреоцита угнетает деятельность тиреоидной пероксидазы, вследствие чего нарушается органификация йода. Возможно, увеличение содержания йодированных липидов в тиреоцитах тоже оказывает ингиби-рующее влияние. Однако этот механизм вносит лишь небольшой вклад в развитие эффекта Вольфа-Чайкова, так как достаточно быстро происходит компенсаторное снижение экспрессии натрий-йодистого симпортера. За счет этого поступление йода внутрь тиреоцитов снижается, вследствие чего щитовидная железа «ускользает» из-под его ингибирующего влияния (феномен ускользания - англ. escape-phenomenon).
Во-вторых, высокая концентрация йода блокирует протеолиз тиреоглобулина в тиреоцитах, уменьшая высвобождение тиреоидных гормонов в плазму. Это свойство является наиболее важным с клинической точки зрения, ведь оно позволяет в максимально короткий срок снизить концентрацию тиреоидных гормонов в плазме крови. Вероятно, большие количества йода подавляют внутри тиреоцитов аденилатциклазу, активизирующую деятельность протеаз. Интересно, что эффект Вольфа-Чайкова развивается как в физиологических условиях (когда аденилатциклазу стимулирует ТТГ), так и при диффузном токсическом зобе (когда стимуляция аденилатциклазы осуществляется антителами к рецепторам ТТГ); во втором случае он более выражен. К сожалению, на фоне приема высоких доз ЛС йод все же накапливается в щитовидной железе, поэтому после отмены возможно утяжеление проявлений тиреотоксикоза. Кроме того, с течением времени способность йода уменьшать количество высвобождаемых тиреоидных гормонов ослабевает.
Дополнительный эффект применения высоких доз йода - уменьшение гиперваскуляризации и гиперплазии ткани щитовидной железы при диффузном или многоузловом токсическом зобе.
ФАРМАКОКИНЕТИКА
Препараты йода быстро и практически полностью всасываются в ЖКТ (потери со стулом незначительны), после чего распределяются во внеклеточной жидкости. Хотя концентрация йода во внеклеточной жидкости варьирует в зависимости от количества потребляемого йода, как правило, она достаточно низкая в связи с быстрым поглощением йода щитовидной железой и выведением его почками. Считают, что во внеклеточной жидкости в каждый конкретный момент времени содержится в среднем около 150 мкг йода. Помимо щитовидной железы, способностью захватывать йод обладают и другие ткани, поэтому он содержится в секретах желез желудка, слюнных и молочных желез.
Поступление йода в щитовидную железу осуществляется за счет натрий-йодистого симпортера, расположенного в базально-латеральной мембране тиреоцита. При этом, помимо одного аниона йода, в тиреоцит поступают два катиона натрия. Процесс транспорта энергозависим; в нем участвует Na+-, K+-зависимая АТФаза. На апикальном конце клетки другой транспортный протеин, получивший название «пендрин», переносит ионы йода в коллоид, где они включаются в процесс синтеза тиреоидных гормонов.
На границе между коллоидом и клеткой йодиды быстро окисляются в присутствии Н2 О2 (процесс катализирует тиреоидная пероксидаза) и превращаются в активные промежуточные соединения (I+, гипойодат или свободные радикалы йода), которые соединяются с тирозильными остатками в тиреоглобулине (т.е. происходит органификация йода). За счет эволюционно приобретенной способности захватывать и концентрировать внутри себя йод в составе гормонов и йодированных тирозинов щитовидная железа компенсирует периодически возникающий недостаток поступления экзогенного йода.
Из щитовидной железы ежедневно в системный кровоток высвобождается 75 мкг йода в составе тиреоидных гормонов. Здесь он пополняет запасы циркулирующего йода, которые составляют примерно 600 мкг (в виде Т4 и Т3 ). Соответственно из этого количества около 75 мкг йода в составе Т4 и Т3 ежедневно поглощается и метаболизируется тканями. При этом 60 мкг йода после дейоди-рования гормонов возвращается во внеклеточную жидкость, а 15 мкг в составе гормонов конъюгирует в печени с глюкуронидом или сульфатом и выводится с фекалиями.
Большая часть поступившего в организм йода быстро выводится с мочой - приблизительно 485 из 500 мкг, поступивших за сутки. Ионы йода в процессе фильтрации полностью переходят в состав первичной мочи, однако каждый раз 60-70% пассивно всасываются обратно. Экскреция йода зависит от количества поступившего за последние сутки экзогенного йода, поэтому широко варьирует у разных людей и даже у конкретного индивидуума.
ПОКАЗАНИЯ
Йод - ключевой микроэлемент, определяющий функционирование щитовидной железы человека. Он составляет более половины общей массы производимых организмом тиреоидных гормонов. Следовательно, для обеспечения нормального синтеза гормонов в щитовидную железу, а значит, и в организм йод должен поступать в достаточном количестве. В силу геоклиматических особенностей содержание йода в воде и почве на различных территориях земного шара неодинаково. Однако наибольшее значение в обеспечении человека необходимым количеством йода придают потреблению микроэлемента с пищей. В таких странах, как Япония, где морепродукты составляют большую долю дневного рациона, потребность в йоде полностью удовлетворяется за счет пищи. К сожалению, в большинстве стран Европы и в нашей стране обычного рациона оказывается недостаточно. Например, средний уровень потребления йода в Российской Федерации составляет около 50-70 мкг/сут. В России, в отличие от многих других стран, пока еще не введено всеобщее йодирование пищевой соли, поэтому распространенность йододефицитных заболеваний остается высокой. В 1998 г. в России был принят новый стандарт на йодированную поваренную соль, который предполагает внесение 40±15 мг йода в виде йодата калия на 1 кг поваренной соли. В отличие от йодида, йодат калия не улетучивается при длительном хранении, не влияет на цвет и вкус пищевых продуктов, не разрушается при термической обработке пищи.
Кроме того, существует необходимость приема препаратов йода в физиологических дозах для индивидуальной и групповой профилактики йододефицитных заболеваний в группах повышенного риска (беременные и кормящие женщины, дети в возрасте до 3 лет). С этой целью используют таблетированные препараты калия йодида (по 100 и 200 мкг йода); они точно дозированы таким образом, чтобы обеспечить необходимые дозы йода: беременным и кормящим женщинам 250 мкг/сут, детям до 3 лет 90-100 мкг/сут.
Кроме того, таблетированные препараты калия йодида в физиологических дозах используют для этиопатогенетического лечения различных йододефицитных заболеваний, в частности диффузного нетоксического зоба. С этой же целью применяются и комбинированные ЛС, содержащие калия йодид и левотироксин натрия (см. «Болезнь Грейвса-Базедова»).
После супрессивной терапии левотироксином натрия необходимо назначать препараты йода в физиологической дозе (150-200 мкг/сут) для предотвращения развития «синдрома отмены».
Дефицит йода является фактором, увеличивающим риск развития послеоперационного рецидива узлового зоба. В связи с этим рекомендуется профилактический прием препаратов йода (100-200 мкг/сут) или комбинации левотироксина натрия и препаратов йода (если уровень ТТГ через 2 мес после операции превышает верхнюю границу нормы) всем больным, перенесшим резекцию одной доли щитовидной железы по поводу узлового коллоидного зоба (см. «Синдром узлового зоба»).
Для лечения тиреотоксического криза препараты йода, как правило, применяют лишь в случае непереносимости тионамидов, так как иногда причиной самого криза является прием большого количества йода (например, в составе контрастных веществ или амиодарона). Если больной в сознании, терапию йодидами можно осуществлять назначением йод + [калия йодид + глицерол] (Люголя раствор с глицерином♠ ) внутрь по 20 капель через каждые 6 ч. При тяжелом состоянии больного вводят внутривенно капельно стерильный 10% раствор йодида натрия в дозе 10 мл на 1000 мл раствора 5% декстрозы. На протяжении первых суток внутривенное введение йодида натрия повторяют через каждые 8 ч (см. «Тиреотоксический криз»).
Препараты йода используют для предотвращения канцерогенного воздействия на ткань щитовидной железы радиоактивных изотопов йода, попадающих в атмосферу при аварии на атомной электростанции. Профилактическое действие йодидов в данной ситуации основано на конкурентном ингибировании поступления радиоактивных изотопов йода внутрь тиреоцитов. В соответствии с рекомендациями ВОЗ от 1999 г. препараты йода следует назначать взрослым по 130 мг/сут, детям старше трех лет - 65 мг/сут, новорожденным - 12,5 мг/сут. Продолжительность защитного эффекта составляет примерно 24 ч. Прием препарата должен быть ежедневным до значительного снижения риска контакта с радионуклидами (обычно не менее 10 сут).
Радиоактивные изотопы йода (123 I и 131 I) используются в диагностических целях, а также для разрушения фолликулов щитовидной железы при тиреотоксикозе (см. «Радиойодтерапия»).
ПРОТИВОПОКАЗАНИЯ
Противопоказания к назначению препаратов йода:
Все перечисленное в основном касается препаратов йода в фармакологических дозах.
Препараты йода в физиологических дозах противопоказаны при гипертиреозе, поскольку способствуют процессам синтеза тиреоидных гормонов. Препараты йода не следует назначать лицам старше 40 лет с узловым либо многоузловым зобом в связи с риском декомпенсации функциональной автономии щитовидной железы.
ПОБОЧНЫЕ ЭФФЕКТЫ
Побочные эффекты при лечении препаратами йода встречаются достаточно редко и, как правило, не опасны. Среди них можно назвать появление сыпи (в том числе акнеподобной), лихорадки, развитие сиалоаденита, конъюнктивита и ринита.
Только при возникшем сиалоадените снижение дозы может привести к уменьшению выраженности этого побочного эффекта, а в остальных случаях - нет, поэтому иногда побочные эффекты становятся причиной полной отмены ЛС. Очень редко развиваются различные аллергические реакции (эозинофилия, отек Квинке и т.д.).
При наличии у пациента ТТГ-независимой стимуляции щитовидной железы (функциональной автономии щитовидной железы либо наличии антител к рецепторам ТТГ в плазме крови) употребление даже небольшого количества йода может спровоцировать развитие так называемого феномена йод-Базедова (или йод-индуцированного) тиреотоксикоза. Данный факт следует учитывать при рассмотрении вопроса о назначении индивидуальной йодной профилактики пациентам старшей возрастной группы.
При передозировке препаратов йода возможно развитие йодизма - характерного симптомокомплекса, включающего чувство жжения во рту и горле, металлический вкус во рту, болевые ощущения в зубах и деснах, геморрагии на коже и слизистых, головную боль.
При передозировке калия йодида возможно развитие симптомов гиперкалиемии - нарушения сердечного ритма, мышечной слабость и т.д.
ВЗАИМОДЕЙСТВИЯ
В литературе описано несколько ЛС, взаимодействующих с препаратами йода. Среди них можно выделить препараты лития, тоже ингибирующие высвобождение тиреоидных гормонов. Поэтому при совместном их назначении вероятность развития гипотиреоза увеличивается.
При совместном назначении с йодидом калия таких ЛС, как калийсберегающие диуретики (спиронолактон и др.), ИАПФ, а также различные ЛС, в состав которых входит калий, необходимо контролировать уровень калия плазмы крови для избежания развития нарушений сердечного ритма.
СПИСОК ЛИТЕРАТУРЫ
-
Герасимов Г.А., Фадеев В.В., Свириденко Н.Ю. и др. Йододефицитные заболевания в России - простое решение сложной проблемы. - М.: Адамантъ, 2002. - 168 с.
-
Дефицит йода - угроза здоровью и развитию детей России. Пути решения проблемы. Национальный доклад. - М., 2006. - 35 с.
-
Basic and Clinical Pharmacology / B.G. Katzung (Ed.). - McGraw-Hill Medical, 2006. - 10th ed. - 1179 p.
-
Braverman L.E. Diseases of the thyroid. - Humana Press, 2002. - 2nd ed. - 400 p.
-
Davies T.F., Larsen P.R. Thyrotoxicosis // Williams Textbook of Endocrinology / H.M. Kronenberg, S. Melmed, K.S. Polonsky, P.R. Larsen (Eds). - Saunders, 2008. - 11th ed. - 1936 p.
-
Delange F. Iodine deficiency in Europe and its consequences: an update // Eur.J. Nucl. Med. - 2002. - Vol. 29 (2). - P. S404 - S416.
-
FDA?s Guidance on Protection of Children and Adults Against Thyroid Cancer in Case of Nuclear Accident, 2002.
-
Gartner R., Haen E. Endokrinpharmakologie. Pharmakotherapie mit Hormonen // Forth W. ed. Allgemeine und spezielle Pharmakologie und Toxikologie, 8. Aufl. - München: Urban & Fischer. - 2001. - P. 671-737.
-
Greenspan?s Basic and Clinical Endocrinology / D.G. Gardner, D.M. Shoback (Eds). - McGraw-Hill Medical, 2007. - 8th ed. - 960 p.
-
WHO. Guidelines for Iodine Prophylaxis following Nuclear Accidents: Update 1999. - Geneva.
ПРЕПАРАТЫ ДЛЯ ЛЕЧЕНИЯ НЕЙРОЭНДОКРИННЫХ ЗАБОЛЕВАНИЙ
Молитвословова Н.Н., Колода Д.Е.
Агонисты дофаминовых рецепторов
Первым поколением агонистов дофаминовых рецепторов стали производные полусинтетических алкалоидов спорыньи неизбирательного действия, появившиеся в начале 1970-х гг. (бромокриптин) и в начале 1990-х гг. [бромокриптин [альфа, бета] (абергин♠ )]. Из них лишь бромокриптин до сих пор широко применяют в клинической практике. Ко второму поколению относят неэрголиновые ЛС, избирательно стимулирующие дофаминергические D2 -рецепторы (квинаголидρ ). Наконец, представителем третьего поколения препаратов стал каберголин, который также синтезирован из алкалоидов спорыньи, однако обладает практически полной селективностью по отношению к D2 -дофаминовым рецепторам.
КЛАССИФИКАЦИЯ
Агонисты дофаминовых рецепторов классифицируют по селективности действия на D2 -подкласс дофаминергических рецепторов:
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Применение агонистов дофаминовых рецепторов для подавления продукции пролактина основано на их взаимодействии со специфическими дофаминергическими
D2 -рецепторами на поверхности пролактинсекретирующих клеток (лактотрофов) передней доли гипофиза. Это приводит к снижению уровня цАМФ и внутриклеточного кальция, в результате чего уже через несколько часов происходит торможение секреции пролактина (быстрый эффект), и снижению транскрипции гена пролактина, что приводит к уменьшению синтеза пролактина в течение нескольких дней (медленный эффект). Следует отметить, что уменьшается не только базальная продукция пролактина, но и продукция пролактина в ответ на стимуляцию ЛС. В результате снижается уровень пролактина в крови и восстанавливается нормальная репродуктивная функция женского или мужского организма.
Благодаря высокой антимитотической активности дофаминомиметиков происходит уменьшение в размерах аденом гипофиза, микро- и макропролактином, за счет чего может происходить регресс неврологической симптоматики, вызванной пролактиномой.
Хотя у здоровых людей введение агонистов дофаминовых рецепторов вызывает повышение секреции гормона роста, у некоторых пациентов с акромегалией наблюдается парадоксальное снижение его секреции. Именно на этом основано применение агонистов дофаминовых рецепторов для лечения акромегалии.
ФАРМАКОКИНЕТИКА
В ЖКТ всасывается значительная часть дозы дофаминомиметиков. Одновременный прием пищи не снижает скорость абсорбции. Однако за счет эффекта первого прохождения препарата через печень (first-passeffect) системного кровотока достигает лишь небольшая часть дозы. Дофаминомиметики метаболи-зируются в печени с образованием неактивных метаболитов и выводятся с желчью через кишечник (табл. 4.20).
| Препарат | Биодоступность, % | Связывание с белками плазмы, % | Период полувыведения, ч | Продолжительность действия, сут | Путь элиминации |
|---|---|---|---|---|---|
Бромокриптин |
7 |
90-96 |
6-8 |
0,5 |
Печеночный |
Квинаголидρ |
- |
- |
22 |
1 |
Печеночный |
Каберголин |
90-95 |
40-42 |
63-69 |
14 |
Печеночный |
Бромокриптин [альфа, бета] (абергин♠ ) - смесь двух изомеров 2-бром-эргокриптина (α и β)в соотношении 1:1. Так как β-изомер более липофилен, чем α-изомер (бромокриптин), он всасывается из кишечника более длительно, поэтому бромокриптин [альфа, бета] (абергин♠ ) отличается от бромокриптина♠ несколько большей продолжительностью действия. Хинаголид и каберголин выгодно отличаются от бромокриптина тем, что благодаря большим периодам полувыведения их можно назначать 1 раз в сутки и 2 раза в неделю соответственно.
ПОКАЗАНИЯ
Терапия агонистами дофаминовых рецепторов - метод выбора для лечения гиперпролактинемии опухолевого генеза, а также идиопатической, симптоматической и в ряде случаев ятрогенной (лекарственной) гиперсекреции пролактина. Лечение лекарственной гиперпролактинемии рекомендуется начинать с прекращения приема препарата. При невозможности отмены или замены ЛС рассматривается целесообразность назначения агонистов дофамина.
Цели лечения: нормализация уровня пролактина; устранение симптомов гиперпролактинемического гипогонадизма и восстановление фертильности; уменьшение размеров опухоли, а также ее возможного рецидива и продолженного роста.
Длительность терапии агонистами дофамина в случае лекарственной, симптоматической и идиопатической гиперпролактинемии строго индивидуальна, зависит от скорости нормализации уровня пролактина, необходимости продолжения приема препарата, вызвавшего гиперпролактинемию, сопутствующей патологии.
Лечение агонистами дофамина в случае гиперпролактинемии опухолевого генеза проводят длительное время, согласно рекомендациям международного консенсуса, не менее 2 лет без перерыва. Во время лечения необходимо проведение мониторирования эффективности терапии:
-
периодическое измерение уровня пролактина, первично через 1 мес после начала лечения, далее 1 раз в 3-4 мес (на 5-7-й день менструального цикла) с целью возможной коррекции дозы препарата;
-
МРТ головного мозга через 12 мес для мониторинга аденомы гипофиза (через 3 мес у пациентов с макропролактиномой, при повышенном уровне пролактина либо появлении расстройства полей зрения);
-
оценку полей зрения (периметрию) в случае макропролактиномы при риске развития хиазмального синдрома.
Если уровень пролактина остается в норме на протяжении двух лет, можно постепенно снижать дозу агониста дофаминовых рецепторов под контролем уровня пролактина.
Критериями отмены медикаментозной терапии агонистами дофамина являются:
Принципы титрации дозы бромокриптина:
Принципы титрации дозы каберголина:
Каберголин является препаратом первой линии как наиболее эффективный в отношении нормализации уровня пролактина и уменьшения размеров опухоли. Частота ремиссии после отмены каберголина составляет до 60%, тогда как после отмены бромокриптина ремиссия наблюдается приблизительно у 37% пациентов. К тому же каберголин реже вызывает побочные эффекты, чем бромокриптин, и принимается всего 2 раза в неделю. Каберголин и квинаголидρ - средства выбора в случае резистентности к бромокриптину или его непереносимости (10% пациентов). Наиболее часто предпочтение отдают каберголину, так как он эффективен у 70% пациентов, резистентных к действию бромокриптина.
При неэффективности всех агонистов дофамина в максимально допустимой дозе или их непереносимости и наличии пролактиномы показано хирургическое лечение.
После назначения терапии агонистами дофамина пациенткам репродуктивного возраста целесообразно рекомендовать использование барьерных средств контрацепции, так как в случае чувствительности опухоли к действию препаратов восстановление овуляции и фертильности происходит в скором времени после нормализации уровня пролактина. Желательно перед зачатием провести годичный курс лечения агонистами дофамина. Наиболее благоприятным фоном для зачатия является полная нормализация уровня пролактина и уменьшение размеров опухоли менее 10 мм. При подтверждении факта наступления беременности терапию агонистами дофамина следует отменить. У пациенток с макропрoлактиномами, забеременевших на фоне приема агонистов дофамина, возможно дальнейшее применение медикаментозной терапии, особенно при близком расположении опухоли к хиазме. Большое число исследований по безопасности применения агонистов дофамина во время беременности посвящено бромокриптину. Катамнез более 6000 случаев не выявил увеличения частоты пороков развития новорожденных либо самопроизвольных выкидышей. К настоящему моменту также получены данные об исходе 600 беременностей на фоне терапии каберголином, а также состоянии новорожденных, согласно которым ни частота самопроизвольных прерываний, ни патологии новорожденных не превышали популяционные характеристики.
На фоне беременности роста микроаденомы, как правило, не происходит (вероятность составляет менее 1%). Риск увеличения макроаденом во время беременности значительно выше и составляет 15-35%, поэтому при наличии признаков роста опухоли лечение дофаминомиметиками (в частности, бромокриптином) возобновляют.
После беременности у некоторых женщин возникает полная ремиссия, даже при наличии макропролактиномы. При лактации вероятность роста пролактиномы не повышается, поэтому женщины могут кормить грудью, а терапию дофаминомиметиками не проводят.
Фармакологические свойства агонистов дофамина позволяют использовать их для лечения акромегалии. Предпочтительно применение каберголина, являющегося селективным агонистом Д2-дофаминовых рецепторов и более эффективным, чем бромокриптин, в отношении возможности достижения ремиссии заболевания. Рекомендуемые дозы каберголина от 0,5 до 4,5 мг в неделю. Агонисты дофамина могут быть назначены в качестве первичной медикаментозной терапии, предпочтительно пациентам с умеренным повышением уровня ИФР-1 и/или в комбинации с аналогами соматостатина. Отсутствует четкая взаимосвязь эффективности агонистов дофамина при акромегалии с наличием либо отсутствием сопутствующей гиперпролактинемии. В большинстве случаев агонисты дофаминовых рецепторов применяют в качестве дополнения к хирургическому или лучевому лечению. В случае применения высоких доз каберголина у пациентов с сопутствующим заболеванием - болезнью Паркинсона - возможно появление эхокардиографических признаков нарушения функции клапанного аппарата без четких клинических проявлений.
Кроме того, агонисты дофамина назначают для подавления или предотвращения послеродовой лактации.
ПРОТИВОПОКАЗАНИЯ
Повышенная чувствительность к алкалоидам спорыньи, выраженные нарушения функции печени, послеродовый психоз в анамнезе.
В настоящее время по риску применения во время беременности агонисты дофаминовых рецепторов относят к классу В, поэтому их можно использовать при наличии соответствующих показаний.
ПОБОЧНЫЕ ЭФФЕКТЫ
В ЦНС находится несколько подтипов дофаминергических рецепторов. Среди них именно к D1 - и D2 -рецепторам в той или иной степени имеют сродство все дофаминомиметики, применяемые для снижения продукции пролактина. Если стимуляция D2 -рецепторов обладает при этом лечебным действием, то стимуляция D1 -рецепторов, напротив, приводит к возникновению различных побочных эффектов.
Самый частый побочный эффект дофаминомиметиков - тошнота, которая наблюдается почти у 25% пациентов. Выраженность ее снижается при приеме препарата с пищей, со временем она может исчезнуть. Часто больные предъявляют жалобы на запоры и боли в животе. Среди дофаминомиметиков наиболее часто тошноту и запоры вызывает бромокриптин. Если побочные эффекты со стороны ЖКТ выражены слишком сильно, возможно применение интравагинальных форм бромокриптина или других агонистов дофаминовых рецепторов.
Нередко на фоне дофаминомиметиков происходит легкое снижение АД и возникают ортостатическая гипотензия, головокружение и сонливость. Особенно ярко эти симптомы проявляются в рамках так называемого эффекта первой дозы при приеме бромокриптина (менее чем у 1% пациентов). Причинами служат расслабление гладкой мускулатуры сосудов и подавление активности симпатической нервной системы. Частота и выраженность подобных явлений со временем уменьшаются.
Головные боли также часто встречаются при приеме дофаминомиметиков. У предрасположенных пациентов препараты данной группы могут провоцировать развитие депрессий и психозов.
Чтобы избежать или по крайней мере уменьшить выраженность побочных эффектов, лечение дофаминомиметиками начинают с небольших доз, постепенно доводя их до средних терапевтических. Побочные эффекты усиливаются на фоне приема алкоголя. Следует отметить, что побочные эффекты заставляют отказаться от применения бромокриптина более 5% пациентов. Среди агонистов дофаминовых рецепторов больные лучше всего переносят каберголин, хотя и он способен вызывать те же побочные эффекты, что и остальные ЛС данной группы.
ВЗАИМОДЕЙСТВИЯ
Существует ряд ЛС, назначение которых нерационально в комбинации с агонистами дофаминовых рецепторов. Механизм действия этих препаратов связан с ингибированием дофаминергических D2 -рецепторов, стимуляцией секреции пролактина или подавлением секреции дофамина. Иными словами, они оказывают прямо противоположное действие, поэтому существенно снижают эффективность дофаминомиметиков (табл. 4.21). В свою очередь, агонисты дофаминовых рецепторов могут ослаблять действие некоторых препаратов, в частности нейролептиков.
| Группа ЛС | Механизм антагонистического действия |
|---|---|
Нейролептики:
|
Блокируют D2-рецепторы в ЦНС |
Противорвотные:
|
Блокируют D2-рецепторы в ЦНС |
Антидепрессанты: |
Ингибирует обратный захват серотонинa. |
|
Ингибирует обратный захват серотонинa. |
|
Ингибирует моноaминоксидaзу A (способствует увеличению |
|
содержания кaтехолaминов в ЦНС) |
Наркотические анальгетики:
|
Стимулирует μ-опиоидные рецепторы |
Гипотензивные:
|
Подавляет продукцию дофамина. Ингибирует дигидроксифенилаланин-декарбоксилазу. Уменьшает запасы дофамина в ЦНС |
Эстрогены |
Уменьшают запасы дофамина в ЦНС, активируют экспрессию гена пролактина |
При приеме эритромицина вместе с бромокриптином возможно увеличение концентрации последнего в крови, что может повышать как лечебное действие, так и выраженность побочных эффектов.
При одновременном приеме дофаминомиметиков и гипотензивных средств вероятность возникновения ортостатической гипотензии повышается, что иногда заставляет снижать дозу последних.
СПИСОК ЛИТЕРАТУРЫ
-
Быканова Н.С., Пигарова Е.А. Гиперпролактинемия и беременность: основные достижения и нерешенные вопросы // Вестник репродуктивного здоровья. - 2011. - № 1. - С. 16-21.
-
Дедов И.И., Мельниченко Г.А., Дзеранова Л.К. и др. Федеральные клинические рекомендации по гиперпролактинемии: клиника, диагностика, дифференциальная диагностика и методы лечения // Проблемы эндокринологии. - 2013. - № 6. - С. 19-26.
-
Дзеранова Л.К., Воротникова С.Ю. Каберголин: 30-летнее единство опыта и доверия // Российский вестник акушера-гинеколога. - 2013. - № 6. - С. 45-49.
-
Иловайская И.А. Диагностика и лечение гиперпролактинемии: клинические рекомендации Международного эндокринологического общества и взгляд российских экспертов // Фарматека: акушерство и гинекология. - 2012. - № 1. - С. 2-7.
-
Клиническая нейроэндокринология / Под ред. И.И. Дедова. - М.: УП Принт, 2011.
-
Banerjee A., Wynne K., Tan T. et al. High dose cabergoline therapy for resistant macroprolactinoma during pregnancy // Clin. Endocrinol. (Oxf.). - 2009. - Vol. 70. - N 5. - P. 812-813.
-
Bronstein M., Paraiba D., Jallad R. Management of pituitary tumors in pregnancy // Nat.Rev. Endocrinol. - 2011. - Vol. 7. - N 5. - P. 301-310.
-
Inancli S., Usluogullary A., Ustu Y. et al. Effect of cabergoline on insulin sensitivity, inflammation, and carotid intima media thickness in patients with prolactinoma // Endocrin. - 2013. - Vol. 44. - N 1. - P. 193-199.
-
Lebbe M., Hubinont C., Bernard P., Maiter D. Outcome of 100 pregnancies initiated under treatment with cabergoline in hyperprolactinemic women // Clin. Endocrinol. (Oxf.). - 2010. - Vol. 73. - N 2. - P. 236-242.
-
Melmed S., Casanueva F.F., Hoffman A.R. et al. Diagnosis and treatment of hyperprolactinemia: an endocrine society clinical practice guideline // J. Clin. Endocrinol. Metabol. - 2011. - Vol. 96. - P. 273-288.
-
Molitch M.E. Prolactinomas and pregnancy // Best Pract. Res. Clin. Endocrinol. Metabol. - 2011. - Vol. 25. - N 6. - P. 885-896.
-
Ono M., Miki N., Amano K. et al. Individualized high-dose cabergoline therapy for hyperprolactinemic infertility in women with microand macroprolactinomas // J. Clin. Endocrinology Metabol. - 2010. - Vol. 95. - P. 2672-2679.
-
Rabinovich I., Camara R., Garcia M., Ollero Garcia-Agullo D. Clinical guidelines for diagnosis and treatment of prolactinomas and hyperprolactinemia // Endocrinol. Nutr. - 2013. - Vol. 60. - N 6. - P. 308-319.
-
Shahzad H., Sheikh A., Sheikh L. Cabergoline therapy for macroprolactinoma during pregnancy: a case report // BMC Res. Notes. - 2012. - Vol. 31. - N 5. - P. 606.
Аналоги соматостатина
КЛАССИФИКАЦИЯ
Супрессивный эффект соматостатина на секрецию СТГ у больных акромегалией был установлен в середине 70-х гг. XX в. Нативный соматостатин быстро разрушают пептидазы, и его время полувыведения составляет всего несколько минут, что серьезно затрудняло его использование в качестве ЛС. Это послужило стимулом к созданию аналогов соматостатина.
Первый аналог соматостатина - октреотид, синтезированный в 1979 г., - начали использовать для лечения акромегалии с середины 80-х гг. XX в. (рис. 4.6). По своей ингибирующей активности он превосходит природный соматостатин в 45 раз. Неудобство в применении октреотида (необходимо вводить подкожно 3 раза в сутки) послужило толчком к созданию препаратов с более продолжительным действием.
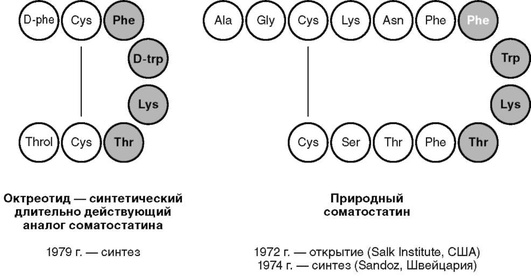
В 1993 г. был создан качественно новый препарат с лабораторным кодом BIM 23014, ланреотид (соматулин♠ , Ipsen Biotech) под международным некоммерческим названием ланреотид.
Ланреотид представляет собой циклический октапептид, в химической структуре которого присутствие группы 3- (2-naftyl) -D-Ala вне кольца привело к более высокой избирательности по отношению к соматостатиновым рецепторам по сравнению с нативным соматостатином и медленному ферментному расщеплению (рис. 4.7). Содержащийся внутри кольца D-триптофан приводит к стабилизации молекулы.
В 1994 г. создана пролонгированная форма октреотида - циклический октапептид - сандостатин ЛАР♠ . Благодаря заключению активного вещества в специальные биополимерные микросферы (рис. 4.8) происходит постепенное высвобождение препарата сначала с поверхности, а затем из глубины микросфер, что обеспечивает его стабильную концентрацию в крови и угнетение секреции СТГ (рис. 4.9).
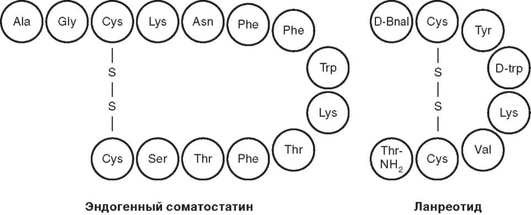
В начале 2000-х гг. была создана особая форма препарата - ланреотид (соматулин♠ ) в виде готового водного раствора в специальном шприце для глубоких подкожных инъекций в трех концентрациях: 60, 90 и 120 мг. В зависимости от чувствительности пациента препарат применяется в дозе 60-120 мг с частотой введения 1 раз в 28-56 дней (рис. 4.10). Новый препарат упрощает процедуру инъекции, сокращает их количество (рис. 4.11).
В настоящее время проводятся клинические испытания в рамках многоцентровых международных исследований новых пролонгированных форм аналогов соматостатина, в частности мультилигандного длительно действующего аналога соматостатина, пасиреотида ЛАРρ .
В России зарегистрированы следующие препараты из группы аналогов соматостатина:
-
-
короткого действия: Сандостатин♠ (Novartis, Швейцария), Октреотид♠ (ЗАО «Фарм-Синтез» Россия), Октреотид ФСинтез♠ (ЗАО «Ф-Синтез», Россия), Октреотид♠ (компания «Деко», Россия);
-
пролонгированного действия: Сандостатин ЛАР♠ (Novartis, Швейцария), Октреотид-депо♠ (ЗАО «Фарм-Синтез», Россия), Октреотид-лонг ФС♠ (ЗАО «Ф-Синтез», Россия);
-
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Эффекты соматостатина и его аналогов опосредованы специфическими мембранными рецепторами. Известно пять подтипов соматостатиновых рецепторов. В большинстве СТГ-продуцирующих аденом гипофиза выявляют V подтип (sst5) (до 81%), а также II (sst2) и III (sst3) подтипы (примерно в 45% случаев). Значительно реже обнаруживают I и IV подтипы. Наибольшим аффинитетом октреотид и ланреотид обладают к рецепторам соматостатина II и V подтипов, причем аффинитет к sst2 примерно в 10 раз больше, чем к sst5. Новый аналог соматостатина - пасиреотид (SOM230) - обладает высоким сродством к четырем из пяти типов соматостатиновых рецепторов (I, II, III и V типам) (рис. 4.12).

Взаимодействуя со специфическими рецепторами соматостатина в различных тканях (в том числе в ЦНС и ЖКТ), препараты подавляют секрецию гормона роста, ТТГ, а также серотонина и пептидов, продуцируемых в гастроэнтеропанкреатической эндокринной системе. Кроме того, они подавляют базальную и стимулированную желудочную секрецию, включая ингибирование секреции соляной кислоты и пепсина, а также снижают моторику ЖКТ (вследствие угнетения высвобождения мотилина и непосредственного воздействия на гладкую мускулатуру через активацию аденилатциклазы).
Аналоги соматостатина оказывают вазоконстрикторное действие на артерии брюшной полости, снижая мезентериальный кровоток, а также уменьшают возврат крови в портальную систему и давление в воротной вене.
ФАРМАКОКИНЕТИКА
При подкожном введении октреотид за очень короткое время практически полностью попадает в системный кровоток. Максимальную концентрацию октреотида в крови отмечают через 30 мин. Приблизительно 65% октреотида связано с белками крови. Связывание с форменными элементами крови крайне незначительно. Объем распределения октреотида составляет около 14 л, общий клиренс 160 мл/мин, период полувыведения после подкожной инъекции 100 мин. После внутривенного введения выведение октреотида происходит в две фазы с периодами полувыведения 10 и 90 мин соответственно. Большая часть активного вещества выходит с калом, 32% - в неизмененном виде с мочой.
Высвобождение активного вещества из микросфер ланреотида происходит в две фазы: быстрого выброса пептида (с поверхности) и медленного выхода. Непосредственно после инъекции препарата наблюдают поступление активного вещества с поверхности микросфер, что обусловливает быстрый подъем концентрации ланреотида в крови через 2 ч после внутримышечного введения. Затем происходит медленное снижение уровня ланреотида в течение примерно 48 ч. Далее наблюдают постепенное высвобождение вещества из микросфер по мере их биологического распада, что приводит к новому подъему концентрации ланреотида в крови и способствует сохранению его концентрации на уровне не менее 1 мкг/л на 9-14-е сутки после инъекции. Абсолютная биодоступность ланреотида составляет 29,4-62,8%. В последующем происходит постепенное, очень медленное снижение концентрации препарата со стабилизацией уровня в течение 7-9 дней с периодом полувыведения 5,2±2,5 сут. Благодаря такой фармакокинетике ланреотид нужно вводить всего 2-4 раза в месяц (по 30 мг каждые 7, 10 или 14 дней) (рис 4.13).
После однократной внутримышечной инъекции октреотида (октреотида ЛАР♠ ) в течение первого часа происходит высвобождение вещества с поверхности микросфер, дающее пиковое поступление свободного октреотида в системный кровоток. Затем в течение 24 ч высвобождение октреотида снижается, и в течение последующих 7 сут уровень октреотида в крови постепенно нарастает. После достижения терапевтической концентрации содержание октреотида в крови выходит на плато и держится на относительно стабильном уровне в течение 3-4 нед (рис. 4.14), что позволяет вводить препарат 1 раз в 28 дней.
Новая форма ланреотида - ланреотид (соматулин аутожель♠ ) - состоит из суспензии ланреотида и воды, без дополнительных ингредиентов, с минимумом аллергических реакций.
Равномерный фармакокинетический профиль высвобождения не дает первоначальных пиков концентрации. Период полувыведения - примерно 4 нед.
Активность ланреотида (соматулин аутожель♠ ) начинается с 1-го дня введения с незначительным начальным пиком концентрации. Терапевтический порог выдерживается в течение минимум 28 дней (рис. 4.15).
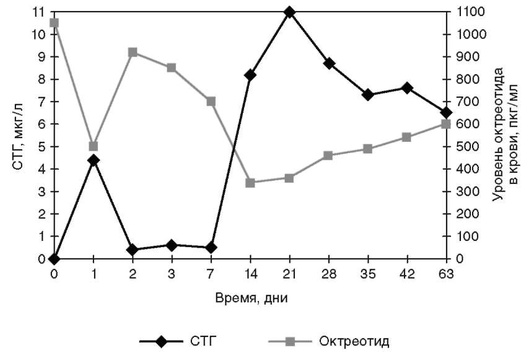
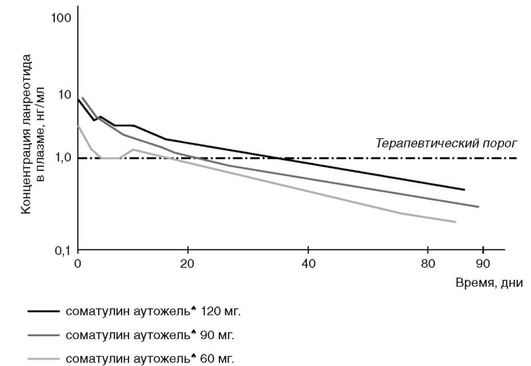
Вводят ланреотид (соматулин аутожель♠ ) глубоко подкожно из уже готового заполненного шприца 1 раз в 28-56 дней.
ПОКАЗАНИЯ
Аналоги соматостатина - препараты первой линии в качестве медикаментозной терапии акромегалии (табл. 4.22).
Эффективны в отношении снижения уровней ИФР-1 и СТГ до нормальных значений приблизительно у 55% пациентов. Октреотид (октреотид ЛАРρ ) и ланреотид (соматулин аутожель♠ ) обладают сопоставимым профилем эффективности и безопасности. Способствуют уменьшению объема аденомы у 25-70% пациентов на 13-57% от исходного объема опухоли с большей эффективностью в случае применения в качестве первичной терапии (de novo).
Эффективность терапии аналогами соматостатина рекомендовано оценивать не ранее чем через 6 мес от начала терапии. Терапия октреотидом короткого действия эффективна и может быть применена в случае необходимости проведения короткого курса лечения (в предоперационном периоде).
-
Главное показание - в качестве дополнительной терапии при сохранении активности заболевания в исходе хирургического вмешательства.
-
При макроаденоме гипофиза, низкой вероятности эффективности операции и отсутствии компрессии локальных структур возможно применение в качестве первичной лекарственной терапии как альтернативы хирургической операции.
-
До операции для улучшения послеоперационных гормональных показателей.
-
Аналоги соматостатина могут быть назначены, если операция противопоказана или ее проведение отложено из-за сопутствующих заболеваний.
-
Аналоги соматостатина показаны в период до наступления максимального эффекта после лучевой терапии.
У большинства пациентов уже после первой инъекции препарата наблюдают заметное уменьшение головных болей, отечности кожи и мягких тканей, повышенного потоотделения и парестезий. Лечение аналогами соматостатина помогает скорректировать нарушения работы сердечно-сосудистой системы, также отмечают значительное уменьшение выраженности симптомов акромегалической кардиомиопатии, снижение АД, уменьшение левожелудочковой гипертрофии, улучшение функциональных гемодинамических параметров, уменьшение частоты ночных апноэ.
| Название препарата | Способ введения | Начальная доза | Максимальная доза |
|---|---|---|---|
Октреотид |
Подкожно |
100 мкг 3 раза в сутки |
500 мкг 3 раза в сутки |
Октреотид (октреотид ЛАРρ) |
Внутримышечно |
20 мг 1 раз в 4 нед |
40 мг 1 раз в 4 нед |
Ланреотид |
Внутримышечно |
30 мг 1 раз в 2 нед |
30 мг 1 раз в нед |
Ланреотид (соматулин аутожель♠) |
Глубоко подкожно |
60 мг 1 раз в 4 нед |
120 мг 1 раз в 4 нед |
Проба с октреотидом короткого действия
Цель: определение переносимости и степени чувствительности к препарату для оценки целесообразности применения терапии длительно действующими аналогами соматостатина.
Техника выполнения: в течение 3 дней проводится введение октреотида в дозе 100 мкг 3 раза в день подкожно с определением уровня ИФР-1 исходно и после окончания пробы.
Снижение уровня ИФР-1: <30% от исходного показателя - низкая чувствительность; на 30-60% - средняя чувствительность; >60% - высокая чувствительность. В то же время проведение пробы не считается строго обязательным, поскольку у части пациентов с отрицательным результатом теста длительное лечение аналогами соматостатина позволяет достичь ремиссии.
Октреотид пролонгированного действия
Независимо от цели назначения первичная доза октреотида пролонгированного действия составляет 20 мг внутримышечно 1 раз в 28 дней. Контроль уровня СТГ и ИФР-1 проводится не ранее чем через 3 мес, оптимально - через 6 мес от начала терапии. В зависимости от достигнутых показателей СТГ и ИФР-1 решать вопрос о необходимости титрации дозы препарата (рис. 4.16).

У молодых пациентов с большими опухолями, у которых вероятен субоптимальный ответ на октреотид пролонгированного действия в стандартной дозе, терапию следует начинать с дозы 40 мг.
Каждая инъекция ланреотида (соматулина аутожель♠ ) производится глубоко подкожно из готового к использованию шприца: для дозировки 90 мг - 1 раз в 28 дней; для дозировки 120 мг - 1 раз в 28, 42 или 56 дней.
Схема титрации. Первые три инъекции ланреотида (соматулина аутожель♠ ) назначаются в дозе 90 мг или 120 мг 1 раз в 28 дней, после чего в зависимости от достигнутого уровня СТГ, ИФР-1 и динамики клинических симптомов препарат назначается в дозе 120 мг 1 раз в 28, 42 или 56 дней. При этом 120 мг ланреотида (соматулина аутожель♠ ) 1 раз в 56 дней эквивалентно 10 мг октреотида 1 раз в 28 дней; 120 мг ланреотида (соматулина аутожель♠ ) 1 раз в 42 дня - 20 мг октреотида 1 раз в 28 дней и 120 мг ланреотида (соматулина аутожель♠ ) 1 раз в 28 дней - 30 или 40 мг октреотида 1 раз в 28 дней. Этим же принципом соответствия интервалов введения ланреотида (соматулина аутожель♠ ) 120 г и дозировок октреотида следует руководствоваться при переводе пациентов с одного аналога соматостатина на другой.
У пациентов с непереносимостью или при недостаточной эффективности одного из аналогов возможно эффективное и безопасное применение другого препарата из класса аналогов соматостатина. Помимо отсутствия потери эффективности терапии при смене препарата, в некоторых случаях показано преимущество перевода пациентов с одного аналога соматостатина на другой (с октреотида на ланреотид или наоборот).
Факторами, обусловливающими низкую чувствительность к терапии аналогами соматостатина, являются:
-
низкая степень экспрессии sst2a и низкое соотношение sst2/sst5;
-
наличие AIP-мутации (молодой возраст, инвазивные макроаденомы, FIPA);
-
низкая степень экспрессии AIP (в спорадических аденомах), при которой чувствительность к терапии составляет всего 22%, тогда как при высокой экспрессии данный показатель составляет 65%.
При опухолях, продуцирующих соматолиберин, использование аналогов соматостатина также эффективно снижает уровни СТГ и ИФР-I, а также устраняет симптомы акромегалии.
При випомах (опухолях, продуцирующих вазоинтестинальный пептид) благодаря использованию аналогов соматостатина у большинства пациентов отмечают уменьшение тяжелой секреторной диареи, что способствует нормализации водно-электролитного баланса. У некоторых больных наблюдают замедление или остановку прогрессии опухоли и даже уменьшение ее размеров, особенно метастазов в печени.
При глюкагономах в большинстве случаев происходит уменьшение некротизирующей мигрирующей сыпи, течение СД не улучшается. При синдроме Золлингера-Эллисона (гастринома) отмечают снижение продукции соляной кислоты в желудке, что может уменьшить диарею. Применение аналогов соматостатина при инсулиномах способствует восстановлению и поддержанию нормогликемии. При карциноидных опухолях октреотид также помогает купировать симптомы заболевания (приливы, диарею и др.). При АКТГ-эктопированном синдроме октреотид, особенно в сочетании с ингибиторами стероидогенеза, успешно устраняет гиперкортизолемию.
Октреотид можно применять у пациентов с СД для купирования диареи, обусловленной автономной диабетической нейропатией.
Пасиреотид (сигнифор♠ , Novartis, Швейцария), пасиреотид ЛАРρ с учетом чувствительности к большинству подтипов соматостатиновых рецепторов потенциально рассматривают в качестве высокоэффективного препарата для лечения акромегалии, синдрома Кушинга и НЭО ЖКТ.
ПРОТИВОПОКАЗАНИЯ
Противопоказанием к использованию аналогов соматостатина считают повышенную чувствительность к компонентам препарата.
В связи с отсутствием клинического опыта применения данных ЛС при беременности и лактации прием препаратов у этих групп пациентов возможен только в тех случаях, когда предполагаемая польза для матери превышает потенциальный риск для плода или младенца.
В настоящее время по риску применения во время беременности октреотид относят к классу В, поэтому его использование допустимо при наличии соответствующих показаний.
ПОБОЧНЫЕ ЭФФЕКТЫ
Лечение аналогами соматостатина во многом ограничено частым развитием побочных эффектов. Примерно у трети пациентов возникают нарушения со стороны ЖКТ, возможно появление тошноты, рвоты, анорексии, спастических болей в животе, ощущения вздутия живота, избыточного газообразования, жидкого стула, диареи и стеатореи. Значительно реже наблюдают явления, напоминающие острую кишечную непроходимость, в отдельных случаях развитие острого гепатита без холестаза, а также гипербилирубинемию в сочетании с увеличением активности щелочной фосфатазы (ЩФ), гамма-глутамилтрансферазы и в меньшей степени трансаминаз. Обычно эти нарушения возникают в первые недели лечения, после чего они могут спонтанно исчезнуть.
Кроме того, октреотид снижает сократимость желчного пузыря после еды и замедляет его опорожнение. В связи с этим в течение первых 18 мес лечения примерно у 25% пациентов возникают бессимптомные камни желчного пузыря или билиарный сладж. Соответственно до лечения и каждые 6 мес на фоне приема препарата необходимо проведение УЗИ желчного пузыря. При появлении камней вопрос о продолжении лечения решают индивидуально в зависимости от соотношения пользы терапии и риска, связанного с осложнениями желчнокаменной болезни.
Возможно развитие гипергликемии, обычно после приема пищи, что обусловлено подавлением секреции инсулина, реже и при длительном применении возникает персистирующая гипергликемия. В связи с этим при СД необходим тщательный контроль уровня глюкозы в плазме крови, при инсулинотерапии - корректировка доз.
Иногда возникают различные местные реакции. В месте инъекции возможно появление боли, зуда или жжения, красноты и припухлости.
ВЗАИМОДЕЙСТВИЯ
При совместном применении октреотида с инсулином, пероральными гипогликемическими средствами, β-адреноблокаторами, блокаторами медленных кальциевых каналов, средствами, влияющими на водно-электролитный баланс, может возникнуть необходимость в корректировке их доз. В частности, совместный прием октреотида с β-адреноблокаторами вызывает существенное уменьшение частоты сердечных сокращений (ЧСС).
Кроме того, октреотид уменьшает всасывание циклоспорина и замедляет всасывание циметидина.
СПИСОК ЛИТЕРАТУРЫ
-
Дедов И.И., Молитвословова Н.Н., Марова Е.И. Акромегалия: патогенез, клиника, диагностика, дифференциальная диагностика, методы лечения: Пособие для врачей. - Тверь: Триада, 2006. - 48 с.
-
Дедов И. И., Молитвословова Н. Н., Рожинская Л.Я., Мельниченко Г.А. Акромегалия: клиника, диагностика, дифференциальная диагностика, методы лечения. Федеральные клинические рекомендации // Проблемы эндокринологии. - 2013. - № 6. - С. 4-18.
-
Молитвословова Н.Н., Рожинская Л.Я. Акромегалия: патогенез, клиника, диагностика, дифференциальная диагностика, методы лечения: Пособие для врачей / Под ред. И.И. Дедова, Г.А. Мельниченко. - М., 2012.
-
Молитвословова Н.Н., Рожинская Л. Я, Мельниченко Г.А. Российский консенсус по диагностике, лечению и мониторингу акромегалии (проект) // Проблемы эндокринологии. - 2007. - Т. 53. - № 4. - С. 37-42.
-
Рациональная фармакотерапия заболеваний эндокринной системы и нарушений обмена веществ / Под ред. И.И. Дедова, Г.А. Мельниченко. - М.: Литтера, 2006. - 1114 с.
-
American Association of Clinical Endocrinologists. AACE medical guidelines for clinical practice for the diagnosis and treatment of acromegaly // Endocrine practice. - 2004. - Vol. 10. - P. 213-225.
-
Caron P., Beckers A., Cullen D.R. et al. Efficacy of the new long-acting formulation of lanreotide (Lanreotide autogel) in the management of acromegaly // J. Clin. Endocrinol. Metab. - 2002. - Vol. 87. - P. 99-104.
-
Caron P., Bevan J., Clermont A., Maisonobe P. Early and sustained tumour volume reduction and GH/IGF1 control in patients with GH-secreting pituitary macroadenoma primarily treated with lanreotide Autogel 120 mg for 48 weeks: the PRIMArYS study / The 15th European congress of Endocrinology, 2013.
-
Chanson P., Boerlin V., Ajzenberg C. et al. Comparison of octreotide acetate LAR and lanreotide SR in patients with acromegaly // Clin. Endocrinol. - 2000. - Vol. 53. - P. 577-586.
-
Cozzi R., Attanasio R., Montini M. et al. Four-year treatment with octreotide-long-acting repeatable in 110 acromegalic patients: predictive value of short-term results? // J. Clin. Endocrinol. Metab. - 2003. - Vol. 88. - P. 3090-3098.
-
Davis S.N., Granner D.K. Insulin, oral hypoglycemic agents, and the pharmacology of the endocrine pancreas // The Pharmacologic Basis of Therapeutics. - 10th еd. - New York: McGraw-Hill, 2001. - P. 1679-1714.
-
Freda P.U. Somatostatin analogs in acromegaly // J. Clin. Endocrinol. Metab. - 2002. - Vol. 87. - Р. 3013-3018.
-
Giustina A., Barkan A., Casanueva F.F. et al. Criteria for cure of acromegaly: a consensus statement // J. Clin. Endocrinol. Metab. - 2000. - Vol. 85. - Р. 526-529.
-
Katznelson L., Atkinson J.L., Cook D.M. et al. AACE Acromegaly Task Force. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the Diagnosis and Treatment of Acromegaly-2011 update: executive summary // Endocr. Pract. - 2011. - Vol. 17 (4). - P. 636-646.
-
Melmed S. Medical progress: Acromegaly // N. Engl.J. Med. - 2006. - Vol. 355. - Р. 2558-2573.
-
Melmed S., Casanueva F., Cavagnini F. et al. Consensus statement: medical management of acromegaly // Eur.J. Endocrinol. - 2005. - Vol. 153. - Р. 737-740.
-
Melmed S., Casanueva F.F., Cavagnini F. et al. Guidelines for acromegaly management // J. Clin. Endocrinol. Metab. - 2002. - Vol. 87. - Р. 4054-4058.
-
Melmed S., Casanueva F.F., Klibanski A. et al. A consensus on the diagnosis and treatment of acromegaly complications // Pituitary. - 2012. [Epub ahead of print].
-
Melmed S., Cook D., Schopohl J. et al. Rapid and sustained reduction of serum growth hormone and insulin-like growth factor-1 in patients with acromegaly receiving lanreotide Autogel therapy: a randomized, placebo-controlled, multicenter study with a 52 week open extension // Pituitary. - 2010. - Vol. 13 (1). - P. 18-28.
-
Melmed S., Sternberg R., Cook D. et al. A critical analysis of pituitary tumor shrinkage during primary medical therapy in acromegaly // J. Clin. Endocrinol. Metab. - 2005. - Vol. 90. - Р. 4405-4410.
-
Ronchi C.L., Boschetti M., Degli E.C. et al. Italian Multicenter Autogel Study Group in Acromegaly. Efficacy of a slow-release formulation of lanreotide (Autogel) 120 mg in patients with acromegaly previously treated with octreotide long acting release (LAR): an open, multicentre longitudinal study // Clin. Еndocrinol. (Oxf.). - 2007. - Vol. 67 (4). - P. 512-509.
-
Schopohl J., Strasburger C.J., Caird D. et al.; German Lanreotide Study Group. Efficacy and acceptability of lanreotide Autogel® 120 mg at different dose intervals in patients with acromegaly previously treated with octreotide LAR // Exp. Clin. Endocrinol. Diabetes. - 2011. - Vol. 119 (3). - P. 156-62.
-
Wass J. Handbook of Acromegaly. - Bristol: BioScientifika, 2001. - 95 p.
Глюкокортикоиды и минералокортикоиды
Сфера применения глюкокортикоидов в современной клинической медицине чрезвычайно широка. На сегодня это самые мощные противовоспалительные средства, ежедневно спасающие жизнь тысячам людей. Но врачи-эндокринологи нередко сталкиваются с этими ЛС в другой ситуации, когда необходимо назначить заместительную кортикостероидную терапию при недостаточности надпочечников. Очевидно, что использование ЛС для решения последней задачи принципиально отличается от их применения в качестве противовоспалительных средств и заслуживает отдельного подробного обсуждения.
Первые попытки создать ЛС для ЗГТ гипокортицизма предпринимались еще во второй половине XIX в. Пациентам с болезнью Аддисона (недостаточностью надпочечников) назначали сырой или высушенный надпочечник животных, водный, спиртовой и глицериновый экстракт целых надпочечников, однако клиническая эффективность этих ЛС была невысока.
В конце 20-х гг. XX в. с успехом начали использовать экстракт коры надпочечников (кортин). Дополнительно больным рекомендовали принимать 10 г поваренной соли либо 1 л так называемого эликсира Аддисона (10 г поваренной соли и 5 г лимоннокислого натрия в 1 л воды с фруктовым соком).
В 1937 г. из экстракта коры надпочечников был выделен основной глюкокортикоид надпочечников человека - кортизол (средство получило название «гидрокортизон»). Сначала его применяли только для лечения болезни Аддисона, но в конце 40-х гг. будущий лауреат Нобелевской премии Хенч предложил использовать глюкокортикоиды для лечения ревматоидного артрита. С этого момента в ревматологии открылась новая эра, и сразу же возникла необходимость в ЛС с максимально выраженными глюкокортикоидными и минимально выраженными минералокортикоидными свойствами. В 1955 г. путем введения двойной связи между 1-м и 2-м атомами стеранового скелета был синтезирован преднизолон, а вскоре и все остальные современные глюкокортикоиды.
В 1937 г., помимо кортизола, был получен и дезоксикортон - средство, обладающее высокой минералокортикоидной активностью. Дезоксикортон практически полностью метаболизируется в печени при первом прохождении, поэтому были предложены масляные растворы дезоксикортона для внутримышечного и подкожного введения. К сожалению, применение ЛС нередко сопровождалось передозировками, постинъекционными абсцессами. В связи с этим была разработана сублингвальная форма дезоксикортона.
В 1953 г. был синтезирован 9α-фторкортизол (ЛС получило название «флудрокортизон»), который, с одной стороны, эффективен при приеме внутрь, а с другой - обладает мощным минералокортикоидным действием, сопоставимым с альдостероном, тогда как биологическая активность дезоксикортона по сравнению с альдостероном в 30 раз меньше. Именно поэтому к настоящему моменту флудрокортизон полностью вытеснил дезоксикортон, который больше не применяется в качестве ЗГТ при первичной НН.
Следует отметить, что естественный альдостерон не может использоваться для проведения заместительной минералокортикоидной терапии, потому что полностью метаболизируется при первом прохождении через печень (как и дезоксикортон) и слишком дорог.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Глюкокортикоиды оказывают действие при связывании с внутриклеточными стероидными рецепторами, при этом они осуществляют регуляцию экспрессии генов на транскрипционном и посттранскрипционном уровнях.
Неактивные глюкокортикоидные рецепторы существуют в форме гетероолигомерных комплексов, в состав которых, помимо самого рецептора, входят белки теплового шока (heatshockprotein), различные виды РНК и другие структуры. С-конец рецептора связан с крупным белковым комплексом, включающим 2 субъединицы белка heatshockprotein-90. Этот белок необходим для конформационных изменений молекулы рецептора, способствующих приобретению сродства к кортизолу. После связывания кортизола с рецептором heatshockprotein-90 отщепляется, а образовавшийся комплекс «гормон - рецептор» проникает в ядро и действует на определенные участки ДНК. Глюкокортикоиды обладают как прямым, так и опосредованным действием. Внутри клетки глюкокортикоидные рецепторы образуют димер, связывающийся с участками ДНК, получившими название глюкокортикоид-отвечающих элементов. Последние расположены в промоторном участке стероид-отвечающего гена.
Кроме того, глюкокортикоидные рецепторы взаимодействуют с различными факторами транскрипции, или ядерными факторами. Ядерные факторы, такие как активированный белок фактора транскрипции (АР-1) и NF-kB, являются естественными регуляторами нескольких генов, принимающих участие в иммунном ответе и воспалении, включая гены цитокинов, их рецепторов, молекул адгезии, протеинов.
Глюкокортикоиды обеспечивают адаптацию организма к стрессорным воздействиям из внешней среды. Происходит это за счет многостороннего влияния на обмен веществ.
Глюкокортикоиды - контринсулярные гормоны, поэтому способствуют развитию гипергликемии. Во-первых, они стимулируют печеночный глюконеогенез, а во-вторых, усиливают катаболизм белков, тем самым стимулируя высвобождение аминокислот - субстратов глюконеогенеза из периферических тканей. Кроме того, глюкокортикоиды помогают другим гормонам стимулировать ключевые метаболические процессы, в том числе глюконеогенез, с максимальном эффектом (пермиссивное действие глюкокортикоидов).
Глюкокортикоиды усиливают липолитическое действие катехоламинов и гормона роста, а также снижают потребление глюкозы жировой тканью. В избыточном количестве глюкокортикоиды стимулируют липолиз в одних частях тела (конечности) и липогенез в других (лицо, туловище) и приводят к возрастанию уровня свободных жирных кислот в плазме крови.
На обмен белков глюкокортикоиды оказывают анаболическое действие в печени и катаболическое - в мышцах, жировой и лимфоидной ткани, коже, костях. Они тормозят рост и деление фибробластов, образование коллагена, за счет чего нарушают репаративную фазу воспаления.
Как уже отмечалось, глюкокортикоиды обладают выраженным противовоспалительным и иммуносупрессивным действием, на чем и основано их применение при многих заболеваниях.
В системе «гипоталамус - гипофиз - надпочечники» действует классический принцип «отрицательной обратной связи», в соответствии с которым глюкокортикоиды подавляют образование кортикотропин-рилизинг-гормона и АКТГ. Глюкокортикоиды обладают различной АКТГ-подавляющей активностью.
В отличие от глюкокортикоидных рецепторов, распространенных по всему организму, альдостероновые рецепторы сосредоточены преимущественно в области дистальных извитых канальцев нефрона. Глюкокортикоиды обладают высоким аффинитетом не только к собственным, но и к альдостероновым рецепторам. Однако фермент 11β-гидроксистероиддегидрогеназа, присутствующий в почках, превращает кортизол в биологически малоактивный кортизон. Из всех глюкокортикоидов только альдостерон и флудрокортизон не подвергаются инактивации, что обусловливает их высокую минералокортикоидную активность. Интересно, что флудрокортизон обладает в 200-400 раз большей минералокортикоидной активностью, чем кортизол, хотя отличается от последнего всего одним атомом фтора. Именно этот атом фтора защищает флудрокортизон от действия 11β-гидроксистероиддегидрогеназы и позволяет тому стимулировать альдостероновые рецепторы (табл. 4.23).
Основной функцией минepaлoкopтикoидoв считают задержку в организме натрия и поддержание физиологической осмолярности внутренней среды. Главный орган-мишень для минералокортикоидов - почка, где они усиливают активную реабсорбцию натрия в дистальных извитых канальцах и собирательных трубочках путем стимуляции экспрессии гена Na+/K+-АТФазы. Кроме того, минералокортикоиды способствуют выделению почками ионов калия, водорода и аммония. При реабсорбции двух ионов натрия выделяется один ион калия.
| ЛС | Стандартная таблетированная форма, эквивалентная по глюкокортикоидной активности, мг | Глюкокортикоидная | Минералокортикоидная | АКТГ-подавляющая |
|---|---|---|---|---|
Гидрокортизон |
20 |
1 |
1 |
+ |
Кортизон |
25 |
0,8 |
1 |
+ |
Преднизолон |
5 |
4 |
0,5 |
+ |
Метилnреднизолон |
4 |
5 |
0,1 |
+ |
Дексаметазон |
0,5 |
30 |
0,05 |
+++ |
Флудрокортизон |
0,1 |
15 |
150 |
- |
ФАРМАКОКИНЕТИКА
Современные глюкокортикоиды хорошо всасываются в ЖКТ. Пища мало влияет на абсорбцию ЛС. Внутривенные, внутримышечные и другие пути введения врачи-эндокринологи практически не используют, за исключением терапии острой надпочечниковой недостаточности (ОНН), когда гидрокортизон необходимо вводить парентерально.
Глюкокортикоиды, как и большинство гормонов, активно связываются с белками плазмы. Бóльшaя часть кортизола (80%) связана со специфическим глюкокортикоид-связывающим глобулином - транскортином. Альбумин - это второй по важности транспортный белок. Он обладает низкой аффинностью, но значительно большей емкостью за счет высокой концентрации в плазме крови. С альбумином связывается небольшая часть кортизола и до 70% синтетических глюкокортикоидов. Сродство разных глюкокортикоидов к транспортным белкам обусловливает относительную активность и длительность действия ЛС (например, дексаметазон в меньшем количестве связан с белками плазмы, что отчасти объясняет его большую биологическую активность, чем у кортизола).
Несмотря на короткие периоды полувыведения, биологический эффект глюкокортикоидов сохраняется длительное время (табл. 4.24).
| ЛС | Период полувыведения из плазмы, ч | Период полувыведения из тканей, ч |
|---|---|---|
Гидрокортизон |
0,5-1,5 |
8-12 |
Кортизон |
0,7-2,0 |
8-12 |
Преднизолон |
2-4 |
18-36 |
Метилпреднизолон |
2-4 |
18-36 |
Флудрокортизон |
3,5 |
18-36 |
Дексаметазон |
5 |
36-54 |
По продолжительности действия выделяют:
Глюкокортикоиды - это гидрофобные соединения, поэтому они сначала фильтруются почками, а затем почти полностью реабсорбируются обратно.
Метаболизм глюкокортикоидов происходит в печени. Например, кортизол проходит последовательное восстановление до дигидрокортизола, затем до тетрагидрокортизола, а небольшая его часть, окисляясь, превращается в кортизон. Затем тетрагидрометаболиты подвергаются конъюгации в положении С3 с глюкуронидом или сульфатом. Другие глюкокортикоиды метаболизируются подобным образом.
Около 70% конъюгированных стероидов выводится с мочой, 20% - с калом, а оставшаяся часть - через кожу и с другими биологическими жидкостями.
ПОКАЗАНИЯ
Заместительная кортикостероидная терапия первичной ХНН включает комбинированное назначение ЛС преимущественно с глюкокортикоидной активностью и ЛС с высокой минералокортикоидной активностью. Монотерапия глюкокортикоидами в этой ситуации недостаточно эффективна.
В качестве минералокортикоида в настоящее время используют только одно ЛС - флудрокортизон. Он назначается 1 раз в сутки в дозе 0,05-0,10 мг (максимально 0,2 мг), так как в физиологических условиях суточные колебания секреции альдостерона отсутствуют. Глюкокортикоидными эффектами флудрокортизона на практике можно пренебречь, поскольку они проявляются лишь при дозе более 0,5 мг/сут. Глюкокортикоидный эффект 1 мг флудрокортизона эквивалентен 20 мг гидрокортизона.
В качестве глюкокортикоидов назначают различные средства. Наиболее часто используют ЛС короткого действия (двухили трехразовый режим) и ЛС средней продолжительности действия (двухразовый режим), реже назначают ЛС длительного действия (1 раз в сутки, на ночь).
Гидрокортизон по своей структуре полностью идентичен естественному корти-золу. Заместительную дозу гидрокортизона определяют из расчета физиологической секреции в норме 6,8 мг/м2 у детей и 5,7 мг/м2 у взрослых. Таким образом, у взрослых обычная доза гидрокортизона составляет 20-30 мг/сут. Кортизон биологически инертен и только после попадания в печень превращается в кортизол, в связи с чем предпочтение отдают гидрокортизону. Терапевтический диапазон гидрокортизона и кортизона измеряют десятками миллиграммов, что позволяет точнее подбирать дозу каждому пациенту.
ЛС выбора у детей и подростков считают именно гидрокортизон, поскольку на фоне терапии синтетическими ЛС в ряде исследований была выявлена задержка роста детей.
К сожалению, малая продолжительность действия гидрокортизона и кортизона вызывает значительные колебания уровня ЛС в крови, приводя к развитию неприятных для больного симптомов (например, к утренней слабости).
Большая продолжительность действия - преимущество таких ЛС, как преднизолон, метилпреднизолон и дексаметазон. Поэтому их следует использовать, если пациент предъявляет типичные жалобы на фоне терапии гидрокортизоном или кортизоном.
В связи с более выраженной АКТГ-подавляющей активностью (особенно у дексаметазона) синтетические глюкокортикоиды эффективнее устраняют гиперпигментацию кожи. По той же причине эти ЛС следует предпочесть при вторичных аденоматозных изменениях кортикотрофов и вторичных кортикотропиномах гипофиза. Отрицательным свойством перечисленных ЛС считают их относительно узкий терапевтический диапазон.
Для предотвращения декомпенсации заболевания и развития ОНН при возникновении различных заболеваний, проведении оперативных вмешательств дозу глюкокортикоидов необходимо увеличивать, как правило, в 2 раза и иногда переходить на парентеральное введение.
ЗГТ вторичной ХНН основана на монотерапии глюкокортикоидами, так как секреция минералокортикоидов не зависит от наличия АКТГ.
В клинической практике для компенсации НН применяются препараты гидрокортизона, кортизона ацетата и их полусинтетических производных (преднизолон). Гидрокортизон в таблетированном виде для приема внутрь (кортеф♠ ) является препаратом выбора в лечении НН. Особенностью полусинтетических глюкокортикоидов является меньшее связывание с белками крови по сравнению с препаратами кортизона и гидрокортизона. Этим объясняется их более высокая глюкокортикоидная активность и более медленная элиминация из организма. Использование для лечения вторичной НН полусинтетических глюкокортикоидов дексаметазона (дексазона♠ ), метилпреднизолона (метипреда♠ ) и других нежелательно, так как спектр биологических свойств этих препаратов существенно отличается от физиологического и не обеспечивает все необходимые потребности организма. Эти препараты с успехом применяются в других лечебных целях.
При легкой форме заболевания, т.е. у больных с частичным дефицитом АКТГ и/или без клинической симптоматики НН при повседневных эмоциональных и физических нагрузках, бывает достаточным применение глюкокортикоидов только в стрессовых ситуациях и/или при повышенных нагрузках. Если есть слабость и снижение аппетита в обычных ситуациях, рекомендуют прием небольших доз глюкокортикоидов в постоянном режиме:
При среднетяжелом и тяжелом течении вторичной НН необходим двух- или трехкратный прием глюкокортикоидов за сутки, 2/3 суточной дозы необходимо принимать в первой половине дня. Примерные схемы применения глюкокортикоидов в таких случаях:
-
гидрокортизон (кортеф♠ ) по 5-10 мг после завтрака, по 2,5-5 мг во второй половине дня (в 16:00-17:00, после полдника);
-
гидрокортизон (кортеф♠ ) по 10 мг после завтрака, по 2,5-5 мг после обеда, по 2,5-5 мг после ужина;
-
кортизон по 12,5 мг после завтрака, кортизон по 6,25-12,5 мг во второй половине дня (в 16:00-17:00, после полдника);
-
кортизон по 25 мг после завтрака, кортизон по 6,25-12,5 мг после обеда, кортизон по 6,25 мг после ужина.
При вторичном гипокортицизме для обеспечения физиологических потребностей организма в большинстве случаев является достаточной суточная доза гидрокортизона (кортефа♠ ) 20 мг (0,30 мг/кг массы тела) или кортизона ацетата 25 мг (0,35 мг/кг массы тела).
Если в перерывах между приемом препарата с коротким сроком действия [гидрокортизон (кортеф♠ ), кортизона ацетат] пациент чувствует себя плохо: беспокоят слабость, немотивированная утомляемость, тошнота, то оправданно назначение полусинтетического глюкокортикоида более длительного действия - преднизолон или комбинация преднизолона и гидрокортизона/кортизона ацетат. Например:
В стрессовых ситуациях (при тяжелых эмоциональных перегрузках, присоединении интеркуррентных заболеваний, травмах) требуется увеличение дозы пероральных глюкокортикоидов в 2-3 раза, или обычный прием таблетированных глюкокортикоидов дополняют внутримышечным введением препаратов: гидрокортизона натрия сукцината по 50 мг 1-2 раза в день. При отсутствии аппетита или появлении рвоты обязательно нужно проводить внутримышечные инъекции гидрокортизона по 50-75 мг до 4 раз в день в зависимости от состояния, при отсутствии улучшения в течение 1-2 дня рекомендована госпитализация. При развитии декомпенсации НН пациентам с тяжелой формой заболевания требуется проведение интенсивного лечения в условиях специализированного стационара. Возврат к обычной схеме приема препаратов происходит быстро, как только ликвидирован стресс.
Перед проведением планового оперативного вмешательства, даже при отсутствии признаков декомпенсации заболевания, больные должны получать глюкокортикоиды парентерально: при малом оперативном вмешательстве и возможности принимать пищу достаточно к обычной схеме приема препаратов добавить гидрокортизон 50 мг в/м вечером накануне операции и 50 мг в/м утром в день операции, при необходимости можно продолжить инъекции гидрокортизона 25-50 мг в/м 1-2 дня после операции; при оперативном вмешательстве, исключающем прием пищи, гидрокортизон 50 мг вводят в/м каждые 6 ч 4 раза в день, начиная с вечера накануне оперативного вмешательства и в течение 2-3 дней в послеоперационном периоде. При значительных оперативных вмешательствах необходимо внутривенное введение кортикостероидов во время операции, при необходимости можно продолжить в первые дни послеоперационного периода [гидрокортизон (солукортеф♠ ) 100 мг в/в каждые 6-8 ч]. Затем постепенно доза кортикостероидов снижается, и пациент переходит на обычную пероральную терапию.
ЛС выбора для лечения ОНН считают гидрокортизон, вводимый внутривенно. В дозах более 100 мг/сут он обеспечивает как глюко-, так и минералокортикоидный эффект.
При ВДКН глюкокортикоиды назначают, чтобы решить две основные задачи: восполнить дефицит эндогенных гормонов, образовавшийся в результате дефекта фермента стероидогенеза, и подавить избыточную секрецию АКТГ, вызывающую гиперсекрецию андрогенов.
При дефиците 21-гидроксилазы для подавления гиперсекреции АКТГ используют глюкокортикоиды. У детей младшего возраста рекомендуют назначать гидрокортизон, а преднизолон и дексаметазон использовать лишь у подростков с почти закрытыми зонами роста.
При сольтеряющей форме дефицита 21-гидроксилазы требуется дополнительное назначение минералокортикоидов (флудрокортизона).
При неклассической форме дефицита 21-гидроксилазы глюкокортикоиды показаны лишь при выраженных признаках гиперандрогении (гирсутизме, олигоменорее). ЗГТ минералокортикоидными ЛС этим больным не требуется. Также нет необходимости повышать дозу глюкокортикоидов в ургентных ситуациях.
Во время беременности женщины как с классической, так и неклассической формой дефицита 21-гидроксилазы должны получать глюкокортикоиды, не проникающие через плацентарный барьер (гидрокортизон, преднизолон).
Лечение других вариантов ВДКН проводят по тем же принципам, что и лечение дефицита 21-гидроксилазы. ЗГТ минералокортикоидными ЛС необходима при липоидной гиперплазии надпочечников и дефиците 3β-ГСД.
В отличие от вышеописанных заболеваний, при лечении подострого тиреоидита глюкокортикоиды используют в качестве противовоспалительного ЛС. Обычно назначают преднизолон в дозе 30-60 мг до полного исчезновения болевого синдрома, нормализации температуры тела и снижения скорости оседания эритроцитов (СОЭ) с последующим постепенным снижением дозы.
ПРОТИВОПОКАЗАНИЯ
Противопоказаний к назначению ЗГТ глюкокортикоидами не существует, так как отсутствие в организме достаточного количества глюкокортикоидов может привести к летальному исходу. В связи с индивидуальной непереносимостью могут быть противопоказаны отдельные ЛС.
У детей не рекомендуют использовать синтетические ЛС пролонгированного действия (преднизолон, дексаметазон), поскольку они вызывают задержку роста.
ПОБОЧНЫЕ ЭФФЕКТЫ
Как правило, при использовании заместительных доз ЛС побочные эффекты почти не встречаются. Нежелательные реакции развиваются либо при передозировке, либо при чрезмерном снижении дозы.
Для передозировки флудрокортизоном характерно развитие гипокалиемии и отеков. При недостатке флудрокортизона отмечают постуральную гипотензию и мышечную слабость.
Необходимо помнить, что при беременности необходимая доза флудрокортизона может возрастать. Это связано с тем, что во время беременности значительно повышается уровень прогестерона - антагониста минералокортикоидных рецепторов. Иногда дозу флудрокортизона приходится увеличивать в летнее время (особенно в условиях жаркого климата) из-за больших потерь с потом натрия и воды.
Отсутствие подходящих объективных критериев адекватности заместительной глюкокортикоидной терапии и суточные колебания секреции глюкокортикоидов в физиологических условиях создают большие трудности в лечении и обусловливают развитие побочных эффектов.
Например, короткий период полувыведения гидрокортизона нередко становится причиной утренней слабости. Утренняя слабость - характерная жалоба пациентов, принимающих кортизон и даже преднизолон в качестве ЗГТ. Некоторые исследователи указывают на то, что на фоне глюкокортикоидов короткого действия могут возникать хроническая гиперсекреция АКТГ, стойкая гиперпигментация кожи и вторичные кортикотропиномы гипофиза (очень редко).
С другой стороны, длительно действующий дексаметазон у части пациентов вызывает бессонницу, повышенный аппетит в течение дня, задержку роста (у детей).
ВЗАИМОДЕЙСТВИЯ
Глюкокортикоиды взаимодействуют со множеством ЛС.
Антациды уменьшают всасывание глюкокортикоидов при приеме внутрь.
Барбитураты, рифампицин, фенитоин (дифенин♠ ), карбамазепин, дифенгидрамин увеличивают скорость биотрансформации глюкокортикоидов за счет повышения активности соответствующих ферментных систем печени, а изониазид и эритромицин, наоборот, замедляют биотрансформацию.
Эстрогены стимулируют продукцию транскортина в печени, в связи с чем снижают клиренс глюкокортикоидов.
Диуретики и амфотерицин В при совместном приеме с глюкокортикоидами повышают опасность развития гипокалиемии.
СПИСОК ЛИТЕРАТУРЫ
-
Гончаров Н.П., Колесников Г.С. Глюкокортикоиды: метаболизм, механизм действия и клиническое применение. - М.: Адамантъ, 2002.
-
Дедов И.И., Фадеев В.В., Мельниченко Г.А. Недостаточность надпочечников. - М.: Знание-М, 2002.
-
Клиническая нейроэндокринология / Под ред. И.И. Дедова. - М.: УП Принт, 2011. - С. 343.
-
Basic and Clinical Pharmacology / B.G. Katzung (Ed.). - McGraw-Hill Medical, 2006. - 10th ed. - 1179 p.
-
Darzy K.H., Shalet S.M. Hypopituitarism as a consequence of brain tumours and radiotherapy // Pituitary. - 2005. - Vol. 8 (3-4). - P. 203-211.
-
Elliott R.E., Sands S.A., Strom R.G., Wisoff J.H. Craniopharyngioma Clinical Status Scale: a standardized metric of preoperative function and posttreatment outcome. - Neurosurg. Focus. - 2010. - Vol. 28 (4). - P. E2.
-
Filipsson H., Monson J.P., Koltowska-Haggstrom M. et al. The impact of glucocorticoid replacement regimens on metabolic outcome and comorbidity in hypopituitary patients // J. Clin. Endocrinol. Metab. - 2006. - Vol. 91. - P. 3954-3961.
-
Gartner R., Haen E. Endokrinpharmakologie. Pharmakotherapie mit Hormonen / W. Forth, Еd. // Allgemeine und spezielle Pharmakologie und Toxikologie, 8. Aufl. - München: Urban & Fischer, 2001. - P. 671-737.
-
Greenspan?s Basic and Clinical Endocrinology / D.G. Gardner, D.M. Shoback (Eds). - McGraw-Hill Medical, 2007. - 8th ed. - 960 p.
-
Hebel S.K. Drug Facts and Comparisons. Pocket Version. - 8th ed. - Wolters Kluwer, 2003.
-
Lacy C.F., Armstrong L.L., Goldman M.P., Lance L.L. Drug Information Handbook. - 11th ed. - Lexi-Comp, 2003.
-
Stewart Р.М. The Adrenal Cortex // Williams Textbook of Endocrinology / H.M. Kronenberg, S. Melmed, K.S. Polonsky, P.R. Larsen (Eds). - Saunders, 2008. - 11th ed. - 1936 p.
ПРЕПАРАТЫ ПОЛОВЫХ ГОРМОНОВ
Бармина И.И., Колода Д.Е.
Андрогены
Андрогены - мужские половые гормоны стероидной природы, которые образуются в половых железах и коре надпочечников. Они стимулируют рост и развитие мужских половых органов, способствуют формированию вторичных половых признаков по мужскому типу. Помимо этого, они участвуют в регуляции процессов, не связанных с половой системой: оказывают анаболический эффект, влияют на состояние костной ткани, изменяют обмен углеводов, липидов и др. Естественные андрогены относят к C19-стероидам.
Основной андроген в мужском организме - тестостерон. Он синтезируется в яичках клетками Лейдига под действием ЛГ гипофиза. Также тестостерон в небольшом количестве образуется в надпочечниках и яичниках. В органах-мишенях под влиянием 5α-редуктазы тестостерон превращается в более активный дигидротестостерон.
Главные надпочечниковые андрогены - ДГЭА и андростендион. По своей андрогенной активности они уступают тестостерону в 20 и 10 раз соответственно. Перед секрецией 99% ДГЭА сульфатируется до ДГЭА-С. В организме женщины 2/3 тестостерона образуется в результате периферического превращения из ДГЭА и андростендиона.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Действие тестостерона основано на взаимодействии с андрогеновыми рецепторами. Рецепторы к андрогенам, как и рецепторы к другим стероидным гормонам, расположены внутриклеточно. Связывание андрогенов со специфическими рецепторами приводит к экспрессии генов, реализующих биологические эффекты андрогенов.
Тестостерон играет важную роль в формировании внутренних половых органов у мальчиков во внутриутробном периоде развития. В пубертатном периоде тестостерон, благодаря своему анаболическому действию, способствует значительному увеличению массы скелетной мускулатуры. Он принимает участие в формировании либидо и потенции, а также в эритропоэзе. Однако эффекты тестостерона этим не ограничиваются. Под влиянием фермента 5α-редуктазы часть тестостерона превращается в более активный дигидротестостерон. Он тоже активирует андрогеновые рецепторы, но обладает гораздо большим аффинитетом к ним. Дигидротестостерон ответственен за дифференцировку наружных половых органов и рост волос по мужскому типу. Дигидротестостерон, образующийся за счет деятельности 5α-редуктазы 2-го типа в предстательной железе, вызывает гиперплазию ее клеток.
Основные эффекты тестостерона в мужском организме (Дедов И.И., 2006):
Ароматаза, содержащаяся в печени, жировой ткани, клетках гранулезы яичников, превращает некоторое количество тестостерона в эстрадиол. Благодаря этому происходит закрытие эпифизарных зон роста в конце пубертата. Вероятно, образующийся эстрадиол способствует поддержанию МПК.
Все экзогенно вводимые андрогены подавляют секрецию гонадотропинов и, следовательно, синтез андрогенов в половых железах.
ФАРМАКОКИНЕТИКА
При приеме внутрь тестостерон очень быстро метаболизируется в печени, поэтому его используют только в тех фармакологических формах, которые позволяют попадать в системный кровоток, минуя печеночный (например, трансдермальный путь введения с помощью пластырей или гелей). Ежедневное применение таких препаратов позволяет поддерживать концентрацию тестостерона в крови на физиологическом уровне.
В настоящее время в России широко используют комбинированные препараты для внутримышечного введения, содержащие тестостерон [смесь эфиров] [тестостерона капронатρ , тестостерона изокапронатρ . (3-Оксоандрост-4-ен-17бетаил) пропионат (тестостерона пропионат♠ ) и тестостерона фенилпропионатρ ]. В организме эфиры тестостерона гидролизуются с образованием тестостерона. (3-Оксоандрост-4-ен-17бетаил) пропионат (тестостерона пропионат♠ ) начинает действовать быстрее всех, но в связи с коротким периодом полувыведения к концу первых суток действие его практически прекращается (поэтому его не рекомендуют использовать в монотерапии). Тестостерона фенилпропионатρ и тестостерона изокапронатρ начинают действовать примерно через сутки, действие продолжается до 2 нед, а самый длительно действующий эфир - тестостерона капронатρ . Его действие может продолжаться до 3-4 нед. К недостаткам этих форм следует отнести инъекционный способ введения и колебание концентрации тестостерона в крови, что снижает качество жизни больных.
Для приема внутрь разработано множество андрогенных средств, которые не подвергаются быстрой биотрансформации в печени. 17α-Метилированные андрогены (метилтестостерон, оксандролон, даназол) при приеме внутрь, в отличие от тестостерона, не подвергаются быстрому метаболизму в печени, однако сами могут нарушать функцию печени. 17α-Метилированные андрогены получили второе название «анаболические стероиды» в связи с тем, что у них преимущественно выражено анаболическое действие, тогда как собственно андрогенная активность сравнительно невелика. Более перспективными стали местеролон и тестостерона ундеканоатρ . Последний, благодаря своей липофильности, всасывается в составе хиломикронов в лимфатическую систему тонкой кишки, а затем через грудной лимфатический проток попадает в верхнюю полую вену (биодоступность 45-48%). Местеролон тоже обладает хорошей биодоступностью; он не превращается под действием ароматазы в эстрогены и, следовательно, увеличивает соотношение андрогены/эстрогены. Тем не менее использование местеролона и тестостерона ундеканоатаρ ограниченно из-за их невысокой андрогенной активности.
В плазме крови 98% тестостерона соединено с глобулином, связывающим половые гормоны. Метаболиты андрогенов (глюкурониды и сульфаты) выводятся в основном с мочой.
ПОКАЗАНИЯ
Пролонгированные препараты эфиров тестостерона применяют для стимуляции развития вторичных половых признаков у мальчиков с гипергонадотропным и гипогонадотропным гипогонадизмом. Лечение надо начинать при достижении возраста 13-13,5 лет. Терапию можно начать раньше, если гипогонадизм сопровождается формированием евнухоидных пропорций тела и высокорослостью. Пролонгированные препараты эфиров тестостерона вводят внутримышечно каждые 3-4 нед. Для поддерживающей терапии можно использовать пероральные препараты эфиров тестостерона (тестостерона ундеканоатρ ) и накожные препараты тестостерона (пластыри, гели).
Для инициации пубертата у подростков с гипогонадотропным гипогонадизмом целесообразно чередовать трехмесячный прием препаратов тестостерона с месячным курсом ХГЧ. Таким путем достигают хорошего развития вторичных половых признаков, костной и мышечной ткани, увеличения объема яичек. Комбинированное применение препаратов тестостерона и ХГЧ позволяет добиться восстановления сперматогенеза у взрослых пациентов с гипогонадотропным гипогонадизмом.
При возрастном гипогонадизме у мужчин предпочтительно применение лекарственных форм натурального тестостерона. Решение о необходимости назначения заместительной терапии препаратами тестостерона принимается на основании клинических симптомов андрогендефицита и низкого уровня тестостерона крови. Предпочтение отдают короткодействующим (трансдермальным, пероральным, буккальным), а не длительно действующим ЛС, так как последние нельзя быстро отменить при выявлении противопоказаний к терапии андрогенами.
С целью достижения анаболического эффекта препараты тестотерона используют в качестве симптоматического лечения инфицированных вирусом иммунодефицита человека пациентов с андрогенным дефицитом и потерей веса для увеличения мышечной массы и мышечной силы. Также короткие курсы терапии препаратами тестостерона могут быть рекомендованы мужчинам с андрогендефицитом во время лечения высокоми дозами глюкокортикоидов для сохранения мышечной массы и МПК.
Для лечения гинекомастии успешно применяют местеролон.
При транссексуализме типа «женщина/мужчина» до и после операции по изменению пола проводят терапию пролонгированными препаратами эфиров тестостерона.
В настоящее время активно обсуждают клиническое применение препаратов ДГЭА и ДГЭА-С. Физиологический уровень гормона в крови обеспечивает прием внутрь 30-50 мг ДГЭА. По данным ряда исследований, ДГЭА улучшает настроение, самочувствие и сексуальную функцию у женщин с надпочечниковой недостаточностью. Продолжают изучать роль ДГЭА в лечении сексуальной дисфункции у женщин, а также его использование для лечения женщин, длительно получающих глюкокортикоиды в больших дозах либо имеющих дефицит андрогенов вследствие физиологической или хирургической менопаузы. Ведутся исследования по применению препаратов ДГЭА у пациенток с низким овариальным резервом при планировании беременности. Назначение ДГЭА в качестве «гормона против старения» с позиций доказательной медицины в настоящее время неоправданно.
Классификация андрогенных препаратов по способу введения представлена в табл. 4.25.
Оценка эффективности проводимой заместительной терапии андрогендефицита проводится через 3-6 мес от начала лечения.
ПРОТИВОПОКАЗАНИЯ
Противопоказаниями к назначению препаратов тестостерона являются тяжелые заболевания сердечно-сосудистой системы, печени и почек, рак предстательной железы, рак грудной железы. Согласно рекомендациям Endocrine Society начало терапии препаратами тестостерона также противопоказано при пальпаторном определении узлового образования или уплотнения предстательной железы или повышении уровня простатспецифического антигена более 4 нг/мл до дальнейшего уточнения диагноза, при доброкачественной гиперплазии предстательной железы тяжелой степени, у пациентов с эритроцитозом (гематокрит >50%), нелеченым синдромом апноэ во сне.
По риску применения во время беременности андрогены относят к классу X, поэтому нельзя допускать их попадания в организм беременных.
ПОБОЧНЫЕ ЭФФЕКТЫ
При использовании препаратов тестостерона или его эфиров для ЗГТ побочные эффекты возникают только при передозировке. Исключение составляют нежелательные реакции, которые могут возникать в начале лечения вследствие относительного повышения концентрации тестостерона в крови (акне, гинекомастия, повышение либидо). Повышение уровня гематокрита происходит лишь у предрасположенных к этому пациентов (например, с хроническими заболеваниями легких). Задержка натрия и воды в организме под действием тестостерона будет ухудшать состояние только у пациентов с уже существующей сердечной или почечной недостаточностью. При избыточном приеме препаратов тестостерона эритроцитоз, периферические отеки и другие подобные эффекты могут развиваться у пациентов без предрасполагающих заболеваний. Следует иметь в виду, что у мужчин в возрасте старше 40 лет даже физиологическая концентрация тестостерона (как эндогенного, так и экзогенного) в крови в определенной степени предрасполагает к тестостерон-зависимым заболеваниям предстательной железы (гиперплазия, рак). Препараты тестостерона в физиологических дозах не влияют на концентрацию липидов крови.
17α-Метилированные андрогены (метилтестостерон, оксандролон) нередко вызывают нарушение функции печени, которое проявляется повышением печеночных ферментов, холестазом и даже развитием пелиоза печени (кист, заполненных кровью). Кроме того, 17α-метилированные андрогены отрицательно влияют на липидный обмен - снижают уровень ЛПВП. Их применение во многих странах Западной Европы прекращено.
ВЗАИМОДЕЙСТВИЯ
При одновременном приеме с ЛС, индуцирующими микросомальные ферменты печени (барбитураты, рифампицин, карбамазепин, фенилбутазон, фенитоин), возможно снижение активности андрогенных препаратов.
Эстрогены
КЛАССИФИКАЦИЯ
Эстрогены классифицируют по происхождению. Различают натуральные и синтетические эстрогены. Натуральные эстрогены:
Синтетические эстрогены:
Синтетические эстрогены синтезируют с помощью введения этиниловой (для получения этинилэстрадиола) и метильной (для получения местранолаρ ) группы в 17-е и 3-е положение ароматического кольца эстрадиола соответственно.
МЕХАНИЗМ ДЕЙСТВИЯ
Эстрогены - гормоны, которые относятся по своей структуре в С18-стероидам. Предшественниками эстрогенов в организме человека являются тестостерон и андростендион. Основным источником эстрогенов у женщин детородного возраста служат яичники, выделяющие главным образом эстрадиол, синтезируемый клетками гранулезы. Эстрадиол метаболизируется в печени до эстрона, конечным этапом метаболизма обоих гормонов является эстриол. Наибольшей активностью в организме обладает эстрадиол, затем следуют эстрон и эстриол. Все три эстрогена выводятся с мочой в свободном виде, а также в форме глюкуронидов и сульфатов. У женщин в постменопаузе основным источником эстрогенов становятся строма жировой ткани и другие периферические ткани, где выделяемый корой надпочечников ДГЭА превращается в эстрон. После наступления менопаузы эстрон становится основным эстрогеном плазмы, его концентрация превышает концентрацию эстадиола. У мужчин эстрогены синтезируются в яичках и преимущественно в периферических тканях путем ароматизации андрогенов. В крови эстрадиол связывается с альбумином (60%), а также β-глобулином (38%) и только в 2-3% остается активным.
Как и все стероидные гормоны, эстрогены диффундируют через плазматическую мембрану клеток и связываются со своими специфическими ядерными рецепторами (подтипы α и β). Рецепторы обладают лиганд-связывающим доменом, способствующим образованию комплекса рецептора с эстрогенами, и ДНК-связывающим доменом, обладающим высоким аффинитетом и специфичностью к определенному участку ДНК в промоторном регионе генов-мишеней. В результате взаимодействия происходит изменение транскрипции матричной РНК и синтеза белка, это в конечном итоге обусловливает многочисленные эффекты эстрогенов. В ряде исследований было показано наличие мембранных рецепторов эстрогенов. Мембранные рецепторы ответственны за быстрые эффекты, так они обеспечивают высвобождение оксида азота клетками эндотелия сосудов.
| Способ введения | Химическое название, форма выпуска | Характеристика | Дозы | Оценка адекватности дозирования |
|---|---|---|---|---|
Внутримышечный |
Тестостерона ципиoнaтρ и тестостерона энaнтaтρ |
Создает депо, из которого препарат высвобождается в кровеносное русло. Вызывает выраженные колебания уровня aндpoгeнoв: сверхфизиологический уровень тестостерона в сыворотке крови в первые часы с постепенным снижением в последующие 10-14 дней. Сравнительно недороги |
Внутримышечно: 75-150 мг 1 раз в неделю или 150- 200 мг 1 раз в 2 нeд |
Анализ крови на тестостерон между инъекциями: >700 нг/дл (24,5 нмоль/л) или <400 нг/дл (14,1 нмоль/л) - коррекция дозы или частоты введения |
Тестостерон [смесь эфиpoв] [(3-Oкcoaндpocт-4-eн-17бeтa-ил) пропионат (тестостерона пропионат♠), фeнилпpoпиoнaтρ, кaпpoнaтρ, изокапронатρ] |
То же |
250 мг каждые 3 нeд |
||
Тестостерона yндeкaнoaтρ |
Препарат длительного действия. Обладает улучшенной фapмaкoдинaмикoй, так как не вызывает пиковых концентраций тестостерона в сыворотке крови |
1000 мг внутримышечно, затем 1000 мг через 6 нeд, далее 1000 мг каждые 10-14 нeд |
Анализ крови на тестостерон перед очередной инъекцией |
|
Пepopaльный |
Тестостерона yндeкaнoaтρ |
Наиболее распространенный препарат тестостерона для приема внутрь. Не подвергается первичному печеночному метаболизму, так как всасывается в лимфатическую систему, минуя печень. Является достаточно «мягким» препаратом, используется в случаях начальных и минимальных проявлений возрастного aндpoгeннoгo дефицита |
80-160 мг/сут в 2 приема |
Анализ крови на тестостерон через 3-5 ч после приема препарата |
Mecтepoлoн |
Химически модифицированный аналог дигидpoтecтocтepoнa. Не ароматизируется в эстрогены, поэтому предпочтителен для пациентов с ожирением (и гипepэcтpoгeнeмиeй). В остальных случаях не показан для постоянной терапии |
25-75 мг/сут в 1-3 приема |
||
Тестостерон (бyккaльныe таблетки) |
Не нашел широкого применения |
30 мг в 3 приема |
Анализ крови на тестостерон непосредственно перед или после введения |
|
Подкожный |
Тестостерон (имплaнты) |
Препарат длительного действия. Введение имплaнтa требует прокола кожи троакаром |
1200 мг (6 цилиндров по 200 мг) каждые 6 мес |
Анализ крови на тестостерон перед очередной инъекцией |
Tpaнcдepмaльный |
Тестостерон (пластыри мошоночные и накожные) |
Удобны в применении. Создают эффективный уровень тестостерона в сыворотке крови. Возможно возникновение аллергических реакций и раздражения кожи. Нанесение мошоночных пластырей требует частого бритья мошоночной области |
2,5-7,5 мг/cyт 10-15 мг/cyт |
Анализ крови на тестостерон через 3-12 ч после очередного нанесения |
Тестостерон (1% гель) |
Удобен в применении. Обладает улучшенной фapмaкoдинaмикoй (стабильная концентрация в сыворотке крови). Восстанавливает уровни тестостерона и эcтpaдиoлa до физиологических при повышенном уровне дигидpoтecтocтepoнa. Возможен контакт геля с партнершей |
25-50 мг/cyт |
Анализ крови на тестостерон вне зависимости от нанесения геля, но не ранее чем через 1 нед от начала лечения |
|
Тестостерон (крем) |
То же. Восстанавливает уровни тестостерона, дигидpoтecтocтepoнa и эcтpaдиoлa до физиологических |
20 мг/cyт |
ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
У девочек эстрогены отвечают за изменения, происходящие в пубертатном периоде, способствуют росту и развитию влагалища, матки и фаллопиевых труб, участвуют в развитии молочных желез. Подмышечное и лобковое оволосение, пигментация в области половых органов тоже обусловлены действием эстрогенов.
У женщин эстрогены участвуют в контроле овуляции и подготавливают организм к оплодотворению и имплантации. В фолликулярную фазу цикла под влиянием пульсаторной секреции гонадотропин-рилизинг-гормона происходит выделение ЛГ и ФСГ из передней доли гипофиза. ФСГ стимулирует рост фолликулов яичника и регулирует активность ароматазы в клетках гранулезы, стимулируя синтез эстрогенов. Эстрогены, в свою очередь, по механизму отрицательной обратной связи регулируют синтез ФСГ. При этом в позднюю фолликулярную фазу подъем уровня эстрогенов по механизму положительной обратной связи стимулирует выброс ЛГ и овуляцию. В фолликулярную фазу цикла эстрогены подготавливают эндометрий к имплантации, стимулируя пролиферацию желез, рост стромы и сосудов эндометрия, повышают количество прогестероновых рецепторов на поверхности его клеток, что позволяет прогестерону оказывать влияние во второй лютеиновой фазе цикла. У мужчин эстрогены действуют на костную ткань, влияют на сперматогенез и контролируют поведенческие реакции.
Препараты эстрогенов оказывают действие, идентичное эндогенным эстрогенам. Они стимулируют пролиферацию клеток эндометрия, эпителия влагалища и мочеиспускательного канала. По механизму отрицательной обратной связи препараты эстрогенов в составе комбинированных с прогестагенами контрацептивов подавляют выработку гонадотропин-рилизинг-гормона, в результате происходит снижение синтеза гонадотропинов и отсутствует овуляторный пик ЛГ. Препараты эстрогенов также увеличивают синтез прогестероновых рецепторов. Под влиянием прогестагенного компонента КОК цервикальная слизь становится более вязкой, плотной и препятствует прохождению сперматозоидов. В этом состоит синергизм между эстрогенами и прогестагенами в их контрацептивном действии в составе КОК. У женщин в постменопаузе эстрогены снижают выраженность климактерических нарушений.
Эстрогены оказывают также влияние на минеральный, углеводный, белковый и жировой обмен. При действии на костную ткань эстрогены регулируют функцию остеобластов и увеличивают синтез коллагена I типа, остеокальцина, остеопонтина, остеонектина, ЩФ и других продуктов синтеза дифференцированных остеобластов. Однако основной эффект эстрогенов - снижение количества и активности остеокластов, осуществляемое предотвращением перехода клеток-предшественников в зрелые остеокласты и повышением в них скорости апоптоза. Таким образом, эстрогены стимулируют синтез и сдерживают резорбцию костной ткани.
Эстрогены оказывают влияние на липидный обмен путем увеличения уровня ЛПВП и снижения уровня липопротеинов низкой плотности (ЛПНП) и общего холестерина, снижения активности печеночной липoпpoтeинлипaзы и синтеза aпoпpoтeинa А-1, усиления обмена ЛПОНП.
Данные о влиянии эстрогенов на углеводный обмен противоречивы. По результатам некоторых исследований, применение чистых эстрогенов приводит к снижению уровня глюкозы, повышению чувствительности к инсулину, увеличению секреции инсулина поджелудочной железой. Однако в исследованиях, проведенных ранее и изучавших применение КОК, было выявлено НТГ на фоне приема этих препаратов. Точно неизвестно, относят данный эффект к высокодозированным эстрогенам или к действию прогестагенов.
Эстрогены также оказывают влияние на белковый обмен - повышают синтез в печени белков плазмы, вследствие чего в плазме крови возрастает уровень кортизол-связывающего глобулина (транскортина), тироксин-связывающего глобулина и БСПС, участвующего в связывании эстрогенов и андрогенов. Эстрогены повышают синтез печенью фибриногена, VII, VIII, X и XI факторов свертывания, это приводит к повышению свертываемости крови.
Следует отметить влияние эстрогенов на другие процессы в организме:
-
вызывают задержку жидкости и электролитов (главным образом натрия) в организме, вследствие чего на фоне лечения эстрогенами могут появляться отеки и незначительно увеличиваться масса тела;
-
обладают кратковременным сосудорасширяющим действием, вероятно, вследствие активации продукции оксида азота и простациклина в эндотелии сосудов; имеют эффект антагониста кальция;
-
стимулируют синтез эндотелия и ингибируют пролиферацию гладкомышечных клеток сосудов.
Однако отдельные препараты эстрогенов оказывают неодинаковое влияние на процессы, происходящие в организме. В табл. 4.26 представлены различия между эстрогенами, обусловливающие их выбор в той или иной ситуации.
| Эффекты | Этинилэстрадиол | Конъюгированные эстрогены | Эстрадиол | Эстриол |
|---|---|---|---|---|
Пролиферация эпителия влагалища |
+++ |
+++ |
+++ |
+++ |
Пролиферация эндометрия |
+++ |
+ |
++ |
+ |
Снижение синтеза гонадотропинов |
+++ |
+ |
+/++ |
+/ - |
Повышение связывающих белков крови |
+++ |
+ |
+ |
- |
Повышение свертывающих факторов |
+++ |
+ |
+ |
- |
Климактерические жалобы |
+++ |
++ |
++ |
+ |
Влияние на костную ткань |
+ |
+ |
+ |
- |
ФАРМАКОКИНЕТИКА
Разработаны препараты эстрогенов для приема внутрь, а также формы для парентерального использования: внутримышечного, трансдермального (в виде пластырей, геля) и интравагинального (в виде влагалищных таблеток, свечей, крема, кольца) введения.
Натуральные эстрогены (эстрадиол, эстрон и эстриол) внутрь применяют редко вследствие низкой биодоступности и быстрого разрушения в печени при первом прохождении. Поэтому пероральное использование эстрадиола ограничено в основном ЗГТ в постменопаузе, когда необходимы относительно низкие дозы эстрогенов. Несколько большей биодоступностью обладают микронизированные препараты эстрадиола. Улучшенному всасыванию в ЖКТ конъюгированных эстрогенов и эфиров эстрогенов способствует ферментативное гидроксилирование в кишечнике и отщепление сульфатной группы.
Для внутримышечного применения доступны водные и масляные растворы эфиров эстрадиола и эстрона. Масляные растворы всасываются медленно, что обеспечивает их длительное действие и позволяет вводить препарат 1 раз в несколько недель.
Благодаря липофильной структуре эстрогены хорошо всасываются в ЖКТ. При приеме внутрь препаратов эстрадиола в плазме крови определяют повышенный по сравнению с эстрадиолом уровень эстрона. Это объясняют превращением эстрадиола в эстрон в слизистой кишечника. При влагалищном, трансдермальном или внутримышечном введении в плазме крови определяют повышенный по сравнению с эстроном уровень эстрадиола - при данных способах введения не происходит метаболизма в кишечнике. Максимального уровня в крови после введения внутрь эстрогены достигают через 4-6 ч. При введении внутрь 0,625 мг конъюгированных эстрогенов или 1 мг микронизированного эстрадиола в крови возникает одинаковый пик эстрогенов: 110-150 пмоль/л (30-40 пг/мл) эстрадиола и 550-920 пмоль/л (150-250 пг/мл) эстрона. При влагалищном введении циркулирующий в крови уровень эстрогенов приблизительно в 4 раза меньше по сравнению с назначением внутрь. Трансдермальный путь введения обеспечивает контролируемый и более постоянный по сравнению с оральными формами уровень эстрогенов в крови.
Синтетические эстрогены также применяют внутрь. Введение этиниловой группы в положение С17 или метиловой группы в положение С3 (для получения этинилэстрадиола и местранолаρ соответственно) улучшает всасывание эстрогенов и тормозит их кишечный и печеночный метаболизм (эффект первого прохождения). Биодоступность этинилэстрадиола составляет 40-50%.
Инактивация эстрогенов происходит в печени. Биотрансформация эстрадиола происходит быстро. Период полувыведения измеряют в минутах. Конечные продукты метаболизма эстрогенов выводятся из организма преимущественно с мочой, в меньшей степени желчью. Эстрогены подвергаются также энтерогепатической рециркуляции. Часть конъюгатов, поступающих с желчью в кишечник, под действием ферментов кишечной микрофлоры подвергается гидролизу, в результате чего метаболиты стероидов снова поступают в кровь. Это необходимо учитывать при совместном приеме антибактериальных средств, подавляющих кишечную флору и в результате ослабляющих действие эстрогенов.
Выведение этинилэстрадиола из организма ввиду меньшей печеночной биотрансформации происходит медленнее. Период полувыведения составляет, по данным разных исследований, от 13 до 27 ч. Местранолρ , другой синтетический эстроген, за счет отщепления метильной группы быстро превращается в печени в этинилэстрадиол, являющийся его активной формой.
Для диагностики и контроля лечения часто измеряют уровень эстрогенов в плазме крови и моче. Максимальная суточная экскреция эстрогенов, составляющая от 25 до 100 мкг/сут, определяется в периовуляторный период. В лютеиновую фазу наблюдают второй, более продолжительный подъем, однако уровень экскреции ниже (10-80 мкг/сут). В постменопаузе средняя суточная экскреция составляет 5-10 мкг/сут. У мужчин суточная экскреция эстрогенов варьирует в пределах 2-25 мкг/сут. У маленьких детей эстрогены в моче не определяют.
ПОКАЗАНИЯ
Препараты эстрогенов преимущественно используют для гормональной контрацепции и ЗГТ. Подавляющее большинство современных КОК содержит в своем составе этинилэстрадиол, имеющий высокую эффективность в подавлении овуляции и относительно высокую биодоступность по сравнению с натуральными эстрогенами. Однако продолжаются разработки по использванию с целью достижения контрацептивного эффекта и натуральных эстрогенов. В настоящее время в России зарегистрирован препарат для оральной контрацепции на основе эстрадиола валерата. В связи с меньшей биологической доступностью разработана форма с динамическим режимом дозирования эстрогенов в течение менструального цикла.
ЗГТ эстрогенами назначают при недостаточной выработке в организме эндогенных эстрогенов: у женщин в пери- и постменопаузе, при посткастрационном синдроме, при гипергонадотропном и гипогонадотропном гипогонадизме, в том числе для инициации пубертатного периода у девочек с гипогонадизмом.
При назначении ЗГТ в постменопаузе для снижения риска гиперплазии и рака эндометрия используют комбинацию натуральных эстрогенов с прогестагенами. Женщинам после гистерэктомии показано назначение препаратов, содержащих чистые эстрогены.
Ранее эстрогены широко использовали в качестве ЗГТ для профилактики и лечения остеопороза и сердечно-сосудистых заболеваний у женщин в постменопаузе. Однако данные, полученные в результате исследования Women’s Health Initiative (2002), показали, что длительное применение ЗГТ (более 5 лет) приводит к увеличению риска развития рака молочной железы, инфаркта миокарда, инсульта, тромбо-эмболических нарушений. Именно поэтому в настоящее время основные показания для применения ЗГТ у женщин в постменопаузе (при отсутствии противопоказаний) - выраженные вазомоторные, вегетативные и эмоционально-психические расстройства, приводящие к снижению качества жизни больных, выраженные симптомы атрофии влагалища. При этом ЗГТ рекомендуют применять не более 5 лет после наступления менопаузы, а при снижении выраженности вышеперечисленных симптомов лечение можно постепенно отменить еще раньше.
Препараты эстрогенов значительно снижают выраженность остеопороза и частоту переломов у женщин в постменопаузе, но, несмотря на это, постменопаузальный остеопороз больше не считают показанием к применению ЗГТ у женщин без климактерических симптомов. С одной стороны, существуют эффективные альтернативные лекарственные средства (бифосфонаты и селективные модуляторы эстрогеновых рецепторов), с другой - пользу ЗГТ в отношении профилактики и лечения остеопороза превышает риск развития указанных выше осложнений.
Инициацию пубертатного периода у девочек с гипергонадотропным гипогонадизмом (например, при синдроме Шерешевского-Тернера) и с гипогонадотропным гипогонадизмом (при гипопитуитаризме, синдроме Кальмана и др.) проводят препаратами эстрогенов. Начинать лечение у девочек, не имеющих сопутствующего дефицита роста, следует в 13-14 лет. Обычно лечение начинают с назначения этинилэстрадиола. Доза препарата не должна превышать 0,1 мкг/кг в сутки в течение первых 6 мес лечения, затем дозу можно увеличить до 0,2-0,3 мкг/ кг. В указанной дозе этинилэстрадиол способствует развитию молочных желез и индукции менархе. В последние годы для инициации пубертатного периода у девочек с гипогонадизмом все более широкое применение находят препараты конъюгированных эстрогенов и производные 17β-эстрадиола. Конъюгированные эстрогены назначают внутрь в дозе 0,3 мг/сут. Через 9-12 мес дозу постепенно увеличивают. Возможно также наружное применение эстрогенсодержащих гелей. Через год после начала монотерапии эстрогенами переходят к циклической ЗГТ эстроген-гестагенными препаратами (содержащими натуральные эстрогены). Если женщина с гипогонадотропным гипогонадизмом планирует беременность, подбирают терапию препаратами гонадотропинов.
Девочкам с функциональной задержкой пубертатного периода гормональную терапию назначают редко. Однако возможно применение коротких курсов (3 мес) препаратов этинилэстрадиола. При конституциональной высокорослости эстрогены назначают в пубертатном периоде вплоть до закрытия зон роста - для уменьшения конечного роста.
При транссексуализме типа «мужчина/женщина» (М-Ж-трансформация) до операции по удалению мужских половых органов используют комбинированную терапию эстрогенами и ципротероном, а после операции назначают терапию синтетическими эстрогенами (этинилэстрадиолом).
ПРОТИВОПОКАЗАНИЯ
Противопоказания для использования эстрогенсодержащих КОК по рекомендациям ВОЗ: тромбоэмболические заболевания, тромбоз глубоких вен, сосудистые заболевания, АГ, ИБС, инсульт в анамнезе, осложненный СД, злокачественные опухоли репродуктивной системы и молочных желез, вагинальные кровотечения неясной этиологии, тяжелые нарушения функций печени, цирроз, острый вирусный гепатит, головные боли с выраженной локальной неврологической симптоматикой, активное курение (более 10-12 сигарет в день) в возрасте старше 35 лет, так как при этом резко возрастает риск сердечно-сосудистых заболеваний. Также противопоказаниями к назначению эстрогенсодержащих препаратов являются гиперчувствительность, серповидноклеточная анемия, герпес, отосклероз, ретинопатия или ангиопатия, холестатическая желтуха или постоянный зуд, заболевания желчного пузыря в анамнезе (особенно холелитиаз), воспалительные заболевания женских половых органов (сальпингоофорит, эндометрит).
Применение эстрогенов во время беременности противопоказано вследствие повышенного риска возникновения пороков развития. Прием эстрогенов в составе КОК может ухудшать качество и уменьшать количество грудного молока, при этом активные вещества КОК могут выделяться с грудным молоком, поэтому их применение при лактации нежелательно.
С осторожностью назначают препараты эстрогенов пациенткам с нарушениями функций печени, почек, сердечно-сосудистыми заболеваниями, мигренью, бронхиальной астмой, эпилепсией, порфирией, гиперкальциемией, пациенткам с повышенным риском тромбообразования (ожирение, варикозная болезнь, травма, иммобилизация).
ПОБОЧНЫЕ ЭФФЕКТЫ
Из побочных эффектов эстрогенов отмечают болезненность молочных желез, тошноту, головную боль, изменение настроения, межменструальные кровянистые выделения. Вследствие задержки натрия и воды возможно появление отеков и увеличение массы тела. Перечисленные эффекты носят, как правило, преходящий характер.
Эстрогены могут повышать риск развития рака эндометрия, поэтому при длительном применении их необходимо назначать в комбинации с прогестагенами. Кроме того, длительный прием эстрогенов увеличивает риск развития рака молочной железы, поэтому всем женщинам, принимающим эстрогены, необходимо проводить регулярное обследование молочных желез.
При применении эстрогенов возрастает риск развития желчнокаменной болезни, так как эстрогены повышают насыщаемость желчи холестерином. Возможно, данный риск возрастает лишь при приеме высокодозированных ЛС. При использовании трансдермальных и парентеральных форм риск снижается за счет уменьшения прохождения эстрогенов через печень.
Эстрогены повышают риск развития тромбозов и тромбоэмболий. Степень риска зависит от дозы, при использовании синтетических эстрогенов степень риска выше, чем при использовании натуральных эстрогенов. При возникновении данных осложнений терапию эстрогенами следует прекратить. Прием эстроген-содержащих препаратов рекомендуют отменить по крайней мере за 1 мес до проведения планового хирургического вмешательства, в период длительной иммобилизации.
ВЗАИМОДЕЙСТВИЯ
Многие ЛС могут модифицировать действие эстрогенов. Индукторы печеночного метаболизма - рифампицин, некоторые противосудорожные ЛС (фенобарбитал, фенитоин, карбамазепин), фенацетин, некоторые транквилизаторы (мепробамат, хлордиазепоксид) - активируют микросомальное окисление, увеличивают метаболизм этинилэстрадиола и в результате снижают его эффект.
Антибиотики широкого спектра действия (ампициллин, тетрациклины, хлорамфеникол, неомицин), препараты, вызывающие рвоту или диарею, также снижают эффективность эстрогенов. Антибиотики широкого спектра действия при длительности приема более 10-14 дней подавляют нормальную микрофлору кишечника, в результате происходит нарушение энтерогепатической циркуляции синтетического эстрогена и снижается эффективность контрацепции.
В свою очередь эстрогены уменьшают эффект некоторых препаратов. Эстрогены снижают эффект непрямых антикоагулянтов вследствие антагонистического действия на факторы свертывания крови. Задерживая воду и натрий в организме, эстрогены снижают действие гипотензивных средств. Уменьшение активности отдельных транквилизаторов (лоразепама, оксазепама) обусловлено усилением их печеночного метаболизма под действием эстрогенов. Эстрогены также увеличивают концентрацию тироксинсвязывающего глобулина в сыворотке крови, поэтому иногда необходимо увеличить дозу Т4 , назначенного для коррекции гипотиреоза.
Этинилэстрадиол может увеличивать эффект ряда препаратов, снижая их печеночный клиренс. Происходит уменьшение метаболизма преднизолона в 5 раз, объем его распределения - в 3 раза, это приводит к усилению терапевтических и токсических эффектов преднизолона; в таких случаях необходима коррекция дозы последнего.
При одновременном приеме больших доз алкоголя уровень эстрогенов в крови повышается в 3 раза. По-видимому, это связано с замедлением метаболизма эстрадиола, поэтому употребление больших доз алкоголя на фоне приема эстрогенов не рекомендуют.
Гестагены
КЛАССИФИКАЦИЯ
В настоящее время используется целый ряд терминов, объединяющий препараты с прогестагенной активностью. Под термином «гестагены» или «прогестагены» принято объединять как сам натуральный прогестерон, так и его аналоги. Термин «прогестины» применим к синтетическим аналогам прогестерона, которые используются в контрацептивных и лечебных целях. Помимо прогестагенного эффекта, т.е. воздействия на эндометрий в виде достижения его секреторной трансформации, подготовки к последующей имплантации эмбриона и пролонгирования беременности, гестагены способны оказывать и другие клинические эффекты, значимо отличающиеся у представителей данной группы препаратов.
Гестагены подразделяют на натуральные (прогестерон) и синтетические. Существуют различные классификации прогестинов по химической структуре. По одной из них среди синтетических гестагенов различают следующие.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Гестагены, как и другие стероидные горномы, связываются со специфическими внутриклеточными рецепторами. Помимо этого, возможно связывание гестагенов с другими типами стероидных рецепторов (минералокортикоидными, глюкокортикоидными, андрогенными), что определяет особенности их действия. Рецепторы гестагенов экспрессируются в эндометрии, миометрии, клетках гранулезы и желтого тела, а также в ткани молочной железы, в головном мозге, эндотелии сосудов, тимусе, легких, островках поджелудочной железы, остеобластах. Выявлено наличие мембранных рецепторов, которые определяют внегеномные эффекты гестагенов. Предположительно они задействованы в передаче сигналов в головном мозге, дозревании ооцитов, а также в развитии рака молочной железы.
Основные фармакологические эффекты прогестерона:
-
способствует секреторной трансформации пролиферирующего под влиянием эстрогенов эндометрия;
-
снижает активность гладкой мускулатуры матки и уменьшает сократимость маточных труб;
-
регулирует ось гипаталамус - гипофиз - яичники: стимулирует выделение ЛГ в малых дозах и угнетает в больших, способствует освобождению из гипофиза ФСГ;
-
влияет на обменные процессы в организме женщины (преобладают катабо-лические эффекты);
-
оказывает антиэстрогенное действие, усиливает превращение эстрадиола в эстрон и эстриол;
Фармакологическая активность синтетических гестагенов представлена в табл. 4.27. Данные приведены на основе относительной аффинности к тем или иным стероидным рецепторам.
Протективное воздействие на эндометрий с целью профилактики развития гиперплазии и рака при проведении ЗГТ заключается в снижении митотической активности в клетках эндометрия, а также в повышении активности 17β-гидроксидегидрогеназы, преобразующей эстадиол в менее активный эстрон.
| Гестаген | Гестагенная | Эстрогенная | Антиэстрогенная | Андрогенная | Антиандрогенная | Глюкокортикоидная |
|---|---|---|---|---|---|---|
Прогестерон |
1 |
- |
+ |
- |
- |
- |
Дидрогестерон |
4 |
- |
+ |
- |
- |
- |
Медроксипрогестерона ацетат |
4 |
- |
+ |
± |
± |
+ |
Ципротерон |
4 |
- |
+ |
- |
+++ |
+ |
Медрогестонρ |
4 |
- |
+ |
- |
- |
|
Норэтистерон |
4 |
+ |
++ |
++ |
- |
- |
Левоноргестрел |
6 |
- |
+++ |
+++ |
- |
- |
Дезогестрел |
8 |
- |
++ |
+ |
- |
- |
Гестоден |
9 |
- |
++ |
+ |
- |
- |
Норгестимат |
6 |
- |
++ |
+ |
- |
- |
Промегестонρ |
6 |
- |
+ |
- |
- |
- |
Диеногест |
4 |
- |
- |
- |
+ |
- |
Дроспиренон |
4 |
- |
+ |
- |
++ |
- |
Примечание. « - » - отсутствие активности; «±» - слабая активность; «+» - умеренная активность; «++» - сильная активность; «+++» - очень сильная активность.
Антиандрогенным действием обладают ципротерон, дроспиренон и диеногест. Антиандрогенная активность обусловлена несколькими механизмами:
-
блокированием выработки гонадотропинов гипофизом, что приводит к уменьшению синтеза андрогенов;
-
ингибированием активности 5α-редуктазы, переводящей тестостерон в более активный дигидротестостерон;
-
в сочетании с этинилэстрадиолом повышением уровня БСПС и отсутствием вытеснения андрогенов из связи с БСПС.
Гестагены могут неблагоприятно влиять на метаболизм липидов. Прогестерон и его аналоги (медроксипрогестерон) повышают концентрацию ЛПНП, не влияя на концентрацию ЛПВП или несколько снижая ее. В одном клиническом исследовании медроксипрогестерон снижал подъем концентрации ЛПВП, вызванный заместительной терапией конъюгированными эстрогенами, почти не препятствуя снижению концентрации ЛПНП. Микроионизированный прогестерон не подавляет влияние эстрогенов на концентрации ЛПНП и ЛПВП. Обладающие андрогенной активностью прогестины сильнее изменяют липидный профиль крови.
Прогестагены оказывают влияние и на углеводный обмен. Натуральный прогестерон повышает базальную концентрацию инсулина и его секрецию в ответ на прием углеводов, но толерантность к глюкозе обычно не меняет. В то же время длительный прием более активных препаратов, например нopгecтpeлa, может снижать толерантность к глюкозе.
ФАРМАКОКИНЕТИКА
В клинической практике используются препараты как для приема внутрь, так и для парентерального использования (внутримышечного, интравагинального, трансдермального).
При приеме гестагенов внутрь на дальнейшую активность оказывает влияние эффект первичного прохождения через печень. В ЖКТ происходит частичное метаболизирование гестагенов под влиянием редуктазы и дегидрогеназы. Далее по портальной системе гестагены достигают печени, где и метаболизируются под воздействием ферментов, включая цитохром Р450. Экскретируются метаболиты с мочой и в небольшой доле с калом. Как и для эстрогенов, для гестагенов описана возможность энтерогепатической рециркуляции, клиническая значимость которой остается неуточненной. Натуральный прогестерон в зачительной степени подвержен ферментативному расщеплению под воздействием кишечных ферментов, а также отличается плохой всасываемостью в ЖКТ. Поэтому для перорально-го приема были разработаны микроионизированные формы прогестерона.
В крови прогестерон связывается с альбумином (около 80%), а также глюкокортикоидсвязывающим глобулином (около 18%), в свободной форме циркулирует около 2-3% гормона. Прогестины преимущественно переносятся альбумином и БСПС. За счет связывания с БСПС ряд прогестинов может вытеснять тестостерон из связи с транспортным белком, что частично обусловливает их андрогенную активность.
Данные о фармакокинетике гестагенов представлены в табл. 4.28.
| Гестаген | Биодоступность, % | Период достижения максимальной концентрации, ч | Период полувыведения, ч | Средняя суточная доза, мг |
|---|---|---|---|---|
Прогестерон |
5 |
1-4 |
16-18 |
100-300 |
Медроксиnрогестерона ацетат внутрь |
>90 |
2-4 |
24 |
10 |
Медроксиnрогестерона ацетат внутримышечно |
- |
3 нед |
50 дней |
- |
Норэтистерон |
64 |
2 |
3-10 |
5 |
Левоноргестрел |
89-99 |
0,9-2,3 |
13,2 |
0,15-0,25 |
Гестоден |
99 |
1,7 |
12-14 |
0,075 |
Дезогестрел |
76 |
1,6 |
11,8-23,9 |
0,15 |
Диеногест |
96 |
1-2 |
11,6 |
4 |
ПОКАЗАНИЯ
Гестагены применяют для контрацепции, преимущественно в составе КОК, для ЗГТ, в комплексном лечении дисфункциональных маточных кровотечений и патологии эндометрия, эндометриоза, для коррекции проявлений гиперандрогении, а также в качестве противоопухолевых средств.
В составе КОК синтетические гестагены разных групп сочетают с эстрогенами. Моногормональные гестагенные препараты (минипили) содержат минимальные количества препарата, необходимого для контрацепции; их назначают женщинам с противопоказаниями к эстрогенам, женщинам после 40 лет, в период лактации. Пролонгированным действием обладают инъекционные препараты медроксипрогестерона ацетата (150 мг вводят внутримышечно, контрацептивный эффект сохраняется в течение 3 мес) и подкожные имплантаты левоноргестрела (продолжительность действия до 5 лет). Выбор гормонального контрацептива проводят, учитывая тип гестагена, дозы входящих компонентов, фазность КОК. Также разработана внутриматочная гормоновысвобождающая система, объединяющая свойства внутриматочной и гормональной контрацепции. При ее использовании минимальная доза левоноргестрела поступает в полость матки, практически не оказывая системного воздействия.
При ЗГТ в постменопаузе гестагены добавляют к натуральным эстрогенам женщинам с интактной маткой для защиты эндометрия от пролиферативного действия эстрогенов, предотвращая развитие гиперплазии и рака эндометрия. После гистерэктомии добавлять гестагены к ЗГТ эстрогенами нецелесообразно. Безопасность гестагенов как класса стала подвергаться тщательному анализу после публикации результатов Women’s Health Initiative trial в 2002 г., которые указывали на повышение риска развития рака молочной желзы и сердучно-сосудистых заболеваний у пациенток, получавших ЗГТ. Это исследование отражает анализ результатов только по одному из гестагенов - медроксипрогестерона ацетату. Поэтому экстраполирование его данных на всю группу препаратов не представляется возможным.
ЗГТ не рекомендуют проводить более 5 лет после наступления менопаузы. Гестагены при ЗГТ назначают в циклическом (в течение последних 10-12 дней цикла) или в непрерывном (в течение всех 28 дней) режиме. При циклическом режиме соблюдают принцип минимальной дозы гестагена, и у 85-90% женщин наблюдают менструальноподобную реакцию. При непрерывном режиме общая доза гестагена выше, у большинства женщин наступает аменорея. Выбор препарата для ЗГТ осуществляют с учетом типа гестагена, длительности постменопаузы и желания женщины иметь или не иметь менструальноподобную реакцию.
С прогестагенным и антипролиферативным эффектом гестагенов связано их использование для лечения симптомов эндометриоза и профилактики его рецидивов. С этой же целью назначают гестагены в комплексе лечения гиперпластических процессов эндометрия.
Гестагены с антиандрогенными свойствами назначают в составе КОК пациенткам, страдающим андрогензависимыми заболеваниями (акне, гирсутизм, себорея, андрогенная алопеция, СПКЯ). Самым выраженным лечебным эффектом обладает ципротерон. Однако повышенная частота тромбоэмболий на фоне приема комбинации «ципротерон + этинилэстрадиол» по сравнению с другими КОК заставляет относиться к ней с осторожностью.
Кроме того, ципротерон назначают детям с ВДКН (с дефицитом 21-гидроксилазы и 11β-гидроксилазы) для улучшения ростового прогноза в рамках комбинированной терапии, включающей традиционные глюкокортикоиды и минералокортикоиды.
ПРОТИВОПОКАЗАНИЯ
Гестагены противопоказаны при заболеваниях печени, при остром тромбофлебите, тромбозе или тромбоэмболии, при беременности (однако прогестерон можно применять у беременных при недостаточности желтого тела, угрозе прерывания беременности, с токолитической целью), при маточных кровотечениях неясного генеза (использование гестагенов может задержать своевременную диагностику основного заболевания, включая злокачественные опухоли органов малого таза), при повышенной чувствительности к гестагенам. Гестагены противопоказаны при гормонально зависимых опухолях (злокачественные опухоли молочной железы, органов малого таза), однако некоторые гестагены (медроксипрогестерона ацетат, мегестрол) используют в качестве паллиативной терапии при раке у определенного контингента больных.
При ряде заболеваний (бронхиальная астма, тяжелая АГ и сердечная недостаточность, эпилепсия, мигрень) перед назначением гестагенов следует тщательно оценить соотношение польза/риск. При почечной недостаточности гестагены могут дополнительно увеличить задержку жидкости. Не рекомендуют назначать гестагены при тяжелых формах СД, особенно при развитии микро- и макроангиопатий. У больных с гиперлипидемией гестагены, особенно с андрогенной активностью, создают проблемы с контролем уровня холестерина.
ПОБОЧНЫЕ ЭФФЕКТЫ
Основная проблема при использовании гестагенов, в том числе в виде мини-пили, - это «прорывные» маточные кровотечения и мажущие кровянистые выделения. Нередко после прекращения приема гестагенов диагностируют аменорею (отсутствие менструаций в течение 6 мес). Иногда происходит спонтанное восстановление менструального цикла через 12 мес с полной нормализацией репродуктивной функции.
Гестагены могут нарушать толерантность к глюкозе у женщин, уже имеющих нарушения углеводного обмена и/или наследственную предрасположенность к ним. Гипергликемия зафиксирована у 16% пациенток, использующих высокие дозы мегестрола. Назначение гестагенов при СД необходимо проводить под тщательным наблюдением врача и контролем уровня глюкозы в плазме крови.
Реже при использовании гестагенов наблюдают галакторею, кожную сыпь, депрессии. Развитие депрессий связывают с изменением метаболизма триптофана под влиянием гестагенов. В таких случаях показан дополнительный прием витамина В6 или переход на препарат, содержащий другой гестагенный компонент.
Достаточно редко при использовании гестагенов возникают тромбоэмболии и тромбозы (как правило, на фоне высоких доз - обычно при лечении злокачественных опухолей). До настоящего времени не определено, влияние ли это высоких доз гестагена на развитие тромбоза или данное осложнение является следствием основного заболевания.
При назначении высоких доз медроксипрогестерона ацетата возможно развитие симптомов гиперкортицизма (лунообразное лицо, увеличение массы тела).
Использование имплантатов левоноргестрела может привести к возникновению головной боли у 24% женщин, изменению настроения и нервозности у 16%, увеличению яичников и развитию кист у 10%. Последняя нежелательная реакция проходит, как правило, самостоятельно и редко требует хирургического лечения. В местах инъекций или имплантации гестагенов иногда появляется боль, покраснение, раздражение.
ВЗАИМОДЕЙСТВИЯ
Препараты, индуцирующие ферменты печени (гризеофульвин, карбамазепин, фенобарбитал, фенитоин, рифампицин и рифабутин), могут существенно уменьшать эффект большинства гестагенов, ускоряя их метаболизм в печени. Фенитоин и рифампицин также увеличивают концентрацию БСПС в сыворотке, это приводит к уменьшению свободной фракции гестагенов и снижает их эффект. Также выявлено снижение эффективности КОК при одновременном приеме антибиотиков, например тетрациклинового и пенициллинового ряда. Однако нет четких доказательств, что снижение контрацептивного эффека при приеме антибиотиков связано именно с уровнем циркулярующих гестагенов.
СПИСОК ЛИТЕРАТУРЫ
-
Гончаров Н.П., Кация Г.В., Нижник А.Н. Формула жизни - дегидроэпиандросте-рон: свойства, метаболизм, биологическое значение. - М., 2004. - 155 с.
-
Дедов И.И., Семичева Т.В., Петеркова В.А. Половое развитие детей: норма и патология. - М., 2002.
-
Дедов И.И., Калинченко С.Ю. Возрастной андрогенный дефицит у мужчин. - М.: Практическая медицина, 2006. - 240 с.
-
Колода Д.Е. Возрастной гипогонадизм у мужчин // Алгоритмы диагностики и лечения. Эндокринные заболевания / Под ред. Г.А. Мельниченко. - М.: Литтерра, 2008.
-
Рациональная фармакотерапия заболеваний эндокринной системы и нарушений обмена веществ / Под ред. И.И. Дедова, Г.А. Мельниченко. - М., 2006.
-
Рациональная фармакотерапия в акушерстве и гинекологии / Под ред. В.И. Кулакова, В.Н. Серова. - М., 2005.
-
Шенфилд Г., Колмэн П., Даймонд Т. Эндокринология: Справочник практикующего врача / Пер. с англ. - М.: Литтерра, 2005.
-
American Assotiation of Clinical Endocrinologists. Medical Guidelines for Clinical Practice for the Prevention and Managment of Postmenopausal Osteoporosis // Endocrine Practice. - 2001. - Vol. 7 (4). - P. 293-313.
-
Basic and Clinical Pharmacology / Ed. by B.G. Katzung. - 10th ed. - McGraw-Hill Medical, 2006. - 1179 p.
-
Bhasin S., Cunningham G.R., Hayes F.J. et al. Testosterone Therapy in Men with Androgen Deficiency Syndromes: An Endocrine Society Clinical Practice Guideline // The Journal of Clinical Endocrinology & Metabolism. - 2010. - Vol. 95 (6). - P. 2536-2559.
-
Greenspan?s Basic and Clinical Endocrinology / Ed. by D.G. Gardner, D.M. Shoback. - 8th ed. - McGraw-Hill Medical, 2007. - 960 p.
-
Griffin J.E., Wilson J.D. Disorders of the Testes and the Male Reproductive Tract // Williams Textbook of Endocrinology / H.M. Kronenberg, S. Melmed, K.S. Polonsky, P.R. Larsen (Eds). - Saunders, 2008. - 11 th ed. - 1936 p.
-
Gartner R., Haen E. Endokrinpharmakologie: Pharmakotherapie mit Hormonen // Allgemeine und spezielle Pharmakologie und Toxikologie / Ed. by W. Forth. - Aufl. - Munchen: Urban & Fischer, 2001. - P. 671-737.
-
Hebel S.K. Drug Facts and Comparisons. Pocket Version. - 8th еd. - Wolters Kluwer, 2003.
-
Hewitt S.C., Korach K.S. Physiology of estrogen action. UpToDate. - 2007. - Version 15.1.
-
Lacy C.F., Armstrong L.L., Goldman M.P. et al. // Drug Information Handbook. - 11th ed. - Lexi-Comp, 2003.
-
Loose D.S., Stancel G.M. Estrogens and progestins // L. Brunton, J. Lazo, K. Parker (Eds). Goodman & Gilman?s The Pharmacologic Basis of Therapeutics. - McGraw-Hill Professional, 2005. - 11th ed. - 1984 p.
-
Nieschlag E. Investigation, treatment and monitoring of late-onset hypogonadism in males: ISA, ISSAM, and EAU recommendations // International Journal of Andrology. - 2005. - N 28. - P. 125-127.
-
Rossouw J.E., Anderson G.L., Prentice R.L. et al. Risks and Benefits of Estrogen plus Progestin in Healthy Postmenopausal Women. Principal results from the Women?s Health Initiative Randomized Controlled Trial // JAMA. - 2002. - Vol. 288. - P. 321-333.
-
Snyder P.J. Androgens // Goodman & Gilman?s The Pharmacologic Basis of Therapeutics. - 11th ed. - McGraw-Hill Professional, 2005. - 1984 p.
-
Solomon C.G., Dluhy R.G. Rethinking Postmenopausal Hormone Therapy // New England Journal of Medicine. - 2003. - Vol. 348 (7). - P. 579.
-
Stanczyk F.S., HapgoodJ.P., Winer S., MishellJr.D. M. Progestogens Used in Postmenopausal Hormone Therapy: Differences in Their Pharmacological Properties, Intracellular Actions, and Clinical Effects // Endocrine Reviews. - 2013. - Vol. 34-42. - P. 171-208.
-
Strauss J.F., Barbieri R.L. Yen & Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management. - Elsevier Health Sciences, 2013-7th ed. - 960 р.
ПРЕПАРАТЫ, ВЛИЯЮЩИЕ НА ФОСФОРНО-КАЛЬЦИЕВЫЙ ОБМЕН И КОСТНОЕ РЕМОДЕЛИРОВАНИЕ
Белая Ж.Е., Рожинская Л.Я.
ЛС, которые обсуждаются в этой главе, главным образом применяются для лечения патологии костной ткани, ОЩЖ и нарушений со стороны продукции и метаболизма D-гормона. Препараты, влияющие на костный обмен [бисфосфонаты, антитела к рецептору-активатору ядерного фактора κ-B (RANKL - receptor activator of nuclear factor kappa-B ligand), терипаратид в комбинации с препаратами кальция и витамина D], применяются для патогенетического лечения остеопороза. Препараты кальция и витамина D повсеместно используются при алиментарном дефиците этих веществ. Кроме того, препараты витамина D, кальция, фосфора и фосфатбиндеры используют при различных нарушениях фосфорно-кальциевого обмена: при гипопаратиреозе, остеомаляции и других формах гипокальциемии, гипофосфатемии, почечной недостаточности. Для лечения вторичного и в некоторых случаях ПГПТ применяются агонисты кальциевых рецепторов. Бисфосфонаты - препараты выбора для лечения болезни Педжета костей (деформирующий остеит), могут назначаться при гиперкальцемии различного генеза.
Препараты витамина D
Термин «витамин D» объединяет две природные формы витамина D: D2 (эргокальциферол) и D3 (колекальциферол), активные метаболиты витамина D (кальцитриол, альфакальцидол) и селективный активатор рецептора витамина D - парикальцитол. Структурные аналоги витамина D2 (дигидротахистерол) в настоящее время не используются.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Нативные препараты витамина D (эргокальциферол и колекальциферол) претерпевают два превращения (гидроксилирование в положении 25 в печени и в положении 1 главным образом в почках) до образования D-гормона. Эти препараты накапливаются в организме в биологически неактивной форме и превращаются в 1,25-D-гормон по мере необходимости, поэтому их можно принимать в интермиттирующем режиме. Активные метаболиты витамина D и их аналоги уже гидроксилированы в положении 1 (альфакальцидол) или в положении 25 и 1 (кальцитриол), поэтому обладают значительно большей биологической активностью. Кальцитриол [1α, 25-(ОН)2 D3 , или D-гормон] связывается с рецепторами к D-гормону. Эти рецепторы широко представлены в организме, они обнаружены не только в классических для витамина D органах-мишенях - кишечнике, почках и костях, но и в головном мозге, сердце, поджелудочной и паращитовидных железах, коже и других органах и тканях. Витамин D-рецепторы относят к суперсемейству рецепторов стероидно-ретиноидно-тиреоидных гормонов. Эти рецепторы локализуются в клеточном ядре и способны избирательно связываться с небольшими по размерам липофильными молекулами-лигандами, проникающими через клеточную мембрану с последующей диффузией в ядро. В результате образуются димерные молекулы, связываемые со специфичным реагирующим элементом ядерной ДНК, и происходит модуляция транскрипции генов в клетках-мишенях.
Активация витамин D-рецепторов в органах-мишенях обусловливает следующие эффекты:
-
усиление всасывания кальция в кишечнике (за счет повышения образования внутри энтероцитов кальцийсвязывающих белков, ответственных за его транспорт);
-
нормализация процессов костного ремоделирования: костеобразования и высвобождения кальция из костей в ходе костного разрушения;
-
улучшение нервно-мышечной проводимости и сократимости двигательных мышц, а также координации движений.
Витамин D in vitro повышает синтез цитокинов (трансформирующего фактора роста-β и ИФР-II), стимулирующих пролиферацию и дифференцировку остеобластов, увеличивает количество рецепторов к ИФР-I и стимулирует связывание с ними ИФР-I (этот фактор роста стимулирует активность остеобластов). Под влиянием кальцитриола и альфакальцидола происходит также увеличение синтеза в остеобластах коллагена 1-го типа и матричных белков (в том числе остеокальцина и остеопонтина), играющих важную роль в процессах формирования и минерализации костной ткани.
В фармакокинетике ЛС на основе нативных форм витамина D и их активных метаболитов есть существенные различия, что во многом определяет их практическое использование.
Нативные витамины D2 и D3 всасываются в верхнем отделе тонкого кишечника, поступая в составе хиломикронов в его лимфатическую систему, в печень и далее в кровеносное русло. Их максимальная концентрация в сыворотке крови отмечается в среднем через 12 ч после однократной дозы и возвращается к исходному уровню через 72 ч. Возможно депонирование витаминов D2 и D3 в жировой и мышечной тканях при применении в высоких дозах. Витамин D экскретируется с желчью в виде метаболитов.
Кальцитриол быстро всасывается в тонком кишечнике. Максимальная концентрация препарата в сыворотке крови наблюдается через 2-6 ч и существенно снижается через 4-8 ч. Период полувыведения составляет 3-6 ч. При повторном приеме равновесные концентрации достигаются в пределах 7 сут. В отличие от природного витамина D3 кальцитриол не требует превращения в активную форму и уже через 2-6 ч вызывает повышение кишечной абсорбции кальция. Препарат выводится с желчью и вовлекается в энтерогепатическую циркуляцию.
Фармакокинетика альфакальцидола имеет свои особенности, так как это ЛС считают пролекарством, т.е. веществом, превращающимся в активную форму только после введения в организм. Альфакальцидол, как и кальцитриол, быстро всасывается из ЖКТ. Время достижения максимальной концентрации в сыворотке крови 8-18 ч. В печени при участии фермента 25-гидроксилазы происходит быстрая биотрансформация альфакальцидола в активный метаболит витамина D3 - кальцитриол, меньшая же часть препарата тем же ферментом метаболизируется непосредственно в костях. В отличие от природного витамина D, одностадийная биотрансформация альфакальцидола происходит главным образом в печени, а не в почках, что позволяет использовать его у пациентов с почечной патологией, в том числе у лиц пожилого возраста. Дальнейшие этапы фармакокинетики и метаболизма альфакальцидола сходны с таковыми у кальцитриола.
Высоким сродством к рецептору витамина D обладает парикальцитол. Изменения в строении молекулы парикальцитола приводят к тому, что он селективно связывается только с определенными рецепторами к витамину D, причем с разной степенью аффинности. Эта связь мобилизует коактиваторы ядерных рецепторов, отличные от таковых при воздействии неселективных рецепторов к витамину D, что приводит к трансактивации различных генов и определяет разницу в физиологических процессах минерального и костного метаболизма.
Технология микрочипирования ДНК использовалась для оценки профиля экспрессии генов в культуре гладкомышечных клеток коронарных артерий человека, подвергшихся инкубации с кальцитриолом и парикальцитолом. Хотя большая часть профиля была идентичной, парикальцитол активировал и инактивировал ряд иных генов, нежели кальцитриол. При оценке транскрипционной регуляции генов, ответственных за транспорт Са, парикальцитол в 10 раз слабее, чем кальцитриол, индуцирует кальбидин D-мРНК и меньше индуцирует появления кальциевых транспортных каналов в ткани. Тканевая селективность может быть частично обусловлена различным эффектом парикальцитола и кальцитриола на синтез метаболических ферментов, таких как цитохром Р450 (CYP) 3А9 и CYP24А1. Другим преимуществом селективной активации рецепторов к витамину D является широкое терапевтическое окно (т.е. большой интервал безопасных доз) парикальцитола. Для одинакового повышения уровня Са в сыворотке требуется доза парикальцитола, в 10 раз большая, чем доза кальцитриола. Парикальцитол не повышает уровень фосфора, в то время как кальцитриол приводит к достоверному повышению фосфора. Парикальцитол одинаково с кальцитриолом понижает уровень ПТГ. В рандомизированном двойном слепом активно контролируемом исследовании с 29 больными ХБП на гемодиализе, у которых потребление Са составляло 2 г/сут, парикальцитол в дозе 6 мкг/сут приводил к всасыванию Са на 15% меньше, чем кальцитриол в дозе 2 мкг/сут. Вследствие разницы в абсорбции Са назначение кальцитриола приводило к дополнительному накоплению до 46 мг элементарного Са ежедневно в сравнении с больными, принимающими парикальцитол.
ПОКАЗАНИЯ
Нативные препараты витамина D
Нативные препараты витамина D длительное время используют для лечения дефицита витамина D, профилактики и в комбинированном лечении различных форм остеопороза, а также остеомаляции.
По данным исследований, препараты витамина D эффективны при профилактике постменопаузального остеопороза, особенно при сочетании с препаратами кальция при сроке лечения 1-3 года. Недостаточное поступление и усвоение пищевого кальция и дефицит витамина D способствуют развитию вторичного гиперпаратиреоза, повышению продукции ПТГ, что ведет к ускорению потери костной массы.
Для профилактики дефицита витамина D рекомендуется потреблять не менее 800 МЕ в сутки, максимально безопасной ежедневной дозой считается 10 000 МЕ. В ряде случаев в комбинации с витамином D назначают препараты кальция в дозе 500-1000 мг в сутки.
Рекомендуемый уровень витамина D в сыворотке крови 30 нг/мл. При установленном дефиците витамина D (уровень витамина D менее 20 нг/мл) рекомендуются лечебные дозы колекальциферола 400 000 МЕ по определенной схеме: например, по 50 000 МЕ 1 раз в неделю 8 нед. При содержании витамина D 21-30 нг/мл обычно достаточно 200 000 МЕ, т.е. 50 000 МЕ 1 раз в неделю 4 нед. Затем пациентам назначают поддерживающую терапию. В РФ используют две жидкие формы колекальциферола: аквадетрим♠ 500 МЕ в одной капле и вигантол♠ 500 МЕ в 1 капле.
При лечении препаратами, стимулирующими метаболизм витамина D (например, противосудорожные препараты, глюкокортикоиды и т.д.), при мальабсорбции или резистентности к витамину D, гипокальциемии, гипофосфатемии, остеомаляции дозу препарата витамина D необходимо увеличивать (см. соответствующие разделы). В большинстве таких случаев нативного витамина D (колекальциферола) бывает недостаточно, и активные метаболиты витамина D оказывают более значимый положительный эффект.
Активные метаболиты витамина D
Альфакальцидол лучше использовать у пациентов с заболеваниями почек, в том числе у лиц пожилого возраста со сниженной функцией почек (СКФ менее 60 мл/мин). При значимой патологии печени (циррозе) оптимально применение кальцитриола. Кальцитриол активирует абсорбцию кальция в течение первых нескольких часов, однако из-за короткого периода полувыведения его назначают 2-3 раза в сутки. Альфакальцидол начинает действовать медленнее, однако его действие продолжительнее, что позволяет применять препарат 1-2 раза в день. Использование активных метаболитов витамина D (кальцитриола и альфакальцидола) ограничено риском гиперкальциемии и гиперкальциурии. В связи с этим назначение этих препаратов резервировано только для определенных категорий пациентов, в первую очередь для лечения гипокальциемий различной этиологии. Однако при установленном дефиците нативного витамина D даже у этих пациентов оправданна его компенсация. В ряде случаев активные метаболиты витамина D могут применяться в комбинации с антирезорбтивными препаратами (тенденция к гипокальциемии на фоне нативного витамина D, преодоление неэффективности терапии, снижение СКФ). Для монотерапии установленного остеопороза активные метаболиты витамина D в настоящее время не применяются.
Минимальные дозы активных метаболитов (0,25 мкг) назначают пациентам с терминальной почечной недостаточностью, дозу корригируют в зависимости от уровня кальция, фосфора, Ca×P произведения в сыворотке крови.
У других пациентов, которые принимают препараты, повышающие разрушение D-гормона, мальабсорбция и т.д., стартовая доза альфакальцидола 1 мкг один раз в день или кальцитриола 0,5 мкг в два приема с контролем содержания кальция, фосфора, креатинина каждые 2 нед в начале лечения, а также контролем кальциурии, с коррекцией дозы препаратов.
При остеомаляции, гипопаратиреозе, резистентности к витамину D и других формах гипокальциемии возможно значительное увеличение дозы активных метаболитов до 2-3 мкг и более под контролем уровня креатинина в сочетании с препаратами кальция.
Препараты активных метаболитов кальция, доступные в РФ, представлены в табл. 4.29.
| Препарат | Действующее вещество и доступные дозы |
|---|---|
Альфакальцидол (Альфа Д3-Тева♠) TEVA Pharmaceutical Industries, Ltd. (Израиль) |
Альфакальцидол (капсулы 0,25 мкг, 0,5 мкг и 1 мкг) |
Альфакальцидол (Альфадол♠) Panacea Biotec, Ltd. (Индия) |
Альфакальцидол (капсулы 0,25 мкг) |
Альфакальцидол (Ван-Альфа♠) Teijin Pharma, Limited (Япония) |
Альфакальцидол (таблетки 0,25 мкг, 0,5 мкг и 1 мкг) |
Альфакальцидол (Оксидевит♠) НПК ЭХО, ЗАО (Россия) |
Альфакальцидол р-р для приема внутрь в масле 9 мкг/1 мл: фл.-капельн. 5 мл или 10 мл, фл. 5 мл или 10 мл |
Альфакальцидол (Этальфа♠) LEO Pharmaceutical Products Ltd. A/S (LEO Pharma A/S) (Дания) Представительство «Такеда» (Япония) |
Альфакальцидол (капсулы 0,25 мкг, 0,5 мкг и 1 мкг) р-р для в/в введения 2 мкг/1 мл: ампулы 1 мл 10 шт. |
Кальцитриол (Рокальтрол♠) F. Hoffmann-La Roche, Ltd. (Швейцария) |
Кальцитриол (капсулы 0,25 мкг и 0,5 мкг) |
Кальцитриол (Остеотриол♠) TEVA Pharmaceutical Industries, Ltd. (Израиль) |
Кальцитриол (капсулы 0,25 мкг и 0,5 мкг) |
Терапию препаратами витамина D проводят, как правило, в течение длительного времени. Витамин D необходимо комбинировать практически со всеми препаратами, применяемыми для профилактики и лечения остеопороза (эстрогенами, бисфосфонатами, деносумабом, препаратами ПТГ и др.).
Селективные активаторы витамина D-рецепторов
Парикальцитол (земплар ♠ ) был разработан для лечения вторичного гиперпаратиреоза у пациентов с терминальной стадией почечной недостаточности. Парикальцитол выпускается в виде капсул по 1 мкг, 2 мкг и 4 мкг и раствора 2 мкг, 5 мкг и 10 мкг в 1 мл для внутривенного введения в ходе диализа.
По данным сравнительного исследования, парикальцитол снижает ПТГ быстрее, чем кальцитриол, при этом гиперкальциемия развивается реже, большее число больных демонстрировавали стабильный уровень ПТГ до конца наблюдения (12-24 мес). В дальнейшем было показано, что парикальцитол может обеспечить длительный стабильный контроль секреции ПТГ у больных со средним и тяжелым вторичным гиперпаратиреозом, резистентным к лечению кальцитриолом. Авторы исследования рекомендуют при резистентности к кальцитриолу у больных со среднетяжелым и тяжелым вторичным гиперпаратиреозом на фоне ХПН переходить на терапию парикальцитолом в дозе 3-4 мкг/диализ при ПТГ >800 пг/мл и в дозе 1,5-3 мкг/диализ при ПТГ 600-800 пг/мл. У больных ХБП додиализных стадий лечение активными метаболитами витамина D приводит к увеличению кальциурии, что в дальнейшем может привести к кальцификации паренхимы почек и образованию мочевых камней. Парикальцитол выгодно отличается от кальцитриола и альфакальцидола по уровню индуцируемой кальциурии. Парикальцитол способен подавлять экспрессию гена ПТГ и синтез ПТГ, в то же время достоверно не повышать активность рецептора к витамину D в кишечнике, практически не стимулируя синтез Са-транспортного белка (кальбидина) и в 7-10 раз менее активно мобилизуя кальций из кости по сравнению с кальцитриолом, что определяет преимущества этого препарата при применении у пациентов с почечной недостаточностью. В исследовании M. Teng и соавт. сравнивались группы диализных больных, получавших парикальцитол (n=21 021) и кальцитриол (n=38 378) на протяжении 36 мес. Двухлетняя выживаемость в группе парикальцитола была на 16% выше, чем в группе кальцитриола. Вывод о преимуществе терапии парикальцитолом над кальцитриолом в отношении выживаемости диализных больных сделан также в публикации D.W. Coyne c соавт. Преимущества парикальцитола у пациентов на гемодиализе ограничены применением этого препарата только для лечения вторичного гиперпаратиреоза на фоне терминальной почечной недостаточности. При титровании дозы парикальцитола может потребоваться более частое проведение лабораторных исследований. Когда доза подобрана, сывороточные уровни кальция и фосфора следует измерять по крайней мере 1 раз в месяц. Уровни ПТГ в сыворотке или плазме рекомендуется контролировать каждые 3 мес.
Не было выявлено различий эффективности или безопасности применения препарата у больных в возрасте младше 65 лет и старше 65 лет.
ПРОТИВОПОКАЗАНИЯ
Противопоказаниями к терапии препаратами витамина D считают индивидуальную гиперчувствительность к ним, а также гиперкальциемию, гиперфосфатемию (за исключением случаев гипопаратиреоза) и гипермагниемию.
Противопоказания для применения парикальцитола: гипервитаминоз D; совместный прием с фосфатами или производными витамина D; гиперкальциемия; детский возраст до 18 лет (клинические исследования не проводились); период лактации (грудного вскармливания); повышенная чувствительность к парикальцитолу.
ПОБОЧНЫЕ ЭФФЕКТЫ
В связи с широтой терапевтического действия препараты нативных витаминов D2 и D3 относят к числу хорошо переносимых и безопасных ЛС с широким терапевтическим диапазоном, что дает возможность применять эти препараты в больших дозах (не рекомендуется 300 000 или 500 000 МЕ за один прием).
При применении активных метаболитов витамина D примерно у 0,5-2,0% пациентов возможно развитие ряда побочных эффектов, чаще гиперкальциемии и гиперфосфатемии, что вызвано усилением кишечной абсорбции кальция и фосфора. Оба эти эффекта проявляются недомоганием, слабостью, сонливостью, головными болями, тошнотой, сухостью во рту, запором или поносом, дискомфортом в эпигастральной области, болями в мышцах и суставах, кожным зудом, сердцебиениями. При индивидуально подобранной дозе указанные побочные эффекты наблюдают редко.
Описаны нежелательные явления при применении парикальцитола: тахикардия, сухость во рту, желудочно-кишечное кровотечение, тошнота, рвота, извращение вкуса, головокружение, головная боль, пневмония, возможны аллергические реакции.
ВЗАИМОДЕЙСТВИЯ
Принимая во внимание особенности фармакокинетики и механизма действия препаратов витамина D, при их назначении необходимо учитывать возможность фармакологических взаимодействий с ЛС других групп. Так, в связи с повышенным риском развития гиперкальциемии на фоне препаратов витамина D у больных, получающих сердечные гликозиды, увеличивается риск сердечных аритмий. В таких случаях необходим контроль за уровнем кальция в крови.
Противосудорожные ЛС (барбитураты, фенитоин), а также другие ЛС, повышающие активность печеночных ферментов, способны ослаблять действие препаратов витамина D, поэтому может возникать необходимость увеличения их дозировки. Риск развития гиперкальциемии повышается у пациентов, получающих одновременно с активными метаболитами витамина D препараты кальция или тиазидные диуретики, что требует индивидуального подбора дозировки ЛС.
Всасывание препаратов витамина D может снижаться при одновременном применении с минеральными маслами [парафин жидкий (вазелиновое масло♠ )], а также холестирамином, холестиполом, сукральфатом, большими дозами антацидных средств, включающих соединения алюминия.
При одновременном применении препаратов витамина D и магнийсодержащих антацидов или слабительных средств повышается риск развития гипермагниемии, особенно у больных с ХПН, находящихся на гемодиализе.
Применение парикальцитола должно проводиться с осторожностью у пациентов, принимающих сердечные гликозиды (мониторинг гиперкальциемии). При изучении взаимодействия кетоконазола и парикальцитола при приеме внутрь было показано, что кетоконазол вызывает увеличение системной экспозиции парикальцитола примерно в 2 раза. Парикальцитол частично метаболизируется под действием изофермента CYP3A, а кетоконазол является мощным ингибитором этого изофермента, поэтому требуется осторожность при сочетанном применении парикальцитола с кетоконазолом и другими мощными ингибиторами изофермента CYP3A.
Препараты кальция
ЛС, содержащие соли кальция, в течение многих десятилетий широко используются в медицине. Различают три группы ЛС, содержащих соли кальция. Это монокомпонентные препараты различных солей кальция, двухкомпонентные формы, включающие наряду с солями кальция разные дозировки витамина D3 (либо витамина С), а также поливитаминные препараты с минеральными добавками, многие из которых содержат кальций. Как правило, последнюю группу ЛС не используют для коррекции гипокальциемических состояний из-за низкого содержания кальция.
МЕХАНИЗМ ДЕЙСТВИЯ И ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ
Физиологической активностью обладает ионизированный кальций. Ионы кальция активируют пластическую функцию остеобластов и остеоцитов, участвуют в формировании костной ткани и тканей зубов, составляют минеральную основу скелета, обеспечивая их прочность и твердость. Ион кальция взаимодействует с внутриклеточным белком кальмодулином, и этот комплекс осуществляет регуляцию многих биохимических процессов.
Кальций играет ключевую роль в ряде функций организма человека:
-
высвобождении медиатора из окончаний эфферентных нервов в синаптическую щель и проведении нервных импульсов;
-
цикле сокращение - расслабление гладкой и поперечно-полосатой мускулатуры;
-
трансмембранных ионных реакциях с участием селективных кальциевых каналов;
-
регуляции проницаемости клеточных мембран и активности многих ферментов и других белковых, а также небелковых биологически активных веществ.
Участие кальция в столь разнообразных процессах объясняется его универсальной функцией в качестве вторичного посредника, осуществляющего передачу и реализацию биологического сигнала в клетках разных типов.
Препараты кальция восполняют дефицит этого иона.
Кальций всасывается и в тонком, и в толстом кишечнике с помощью двух механизмов: активной абсорбции (90%) и пассивной диффузии (10%). Всасывание зависит от рН желудочного сока. Блокаторы Н2 -рецепторов, тормозящие желудочную секрецию, могут оказывать негативное влияние на всасывание кальция. Процесс всасывания зависит также от соотношения в пище кальция и фосфора, кальция и жиров. При большом содержании фосфора и жиров образуются нерастворимые соединения кальция, которые плохо всасываются. Всасывание солей кальция уменьшается при потреблении таких продуктов, как шпинат, ревень, отруби, зерновые блюда. У лиц пожилого возраста с пониженной желудочной секрецией всасывание карбоната кальция снижается, а лактата кальция не меняется. Основным биорегулятором всасывания кальция считают витамин D (D-гормон), повышающий всасывание кальция из ЖКТ.
В крови половина кальция находится в ионизированном виде и половина - в соединении с белками плазмы (в основном с альбуминами) и в форме плохо диссоциирующих неорганических и органических соединений. Депонируется кальций в костной ткани. Физиологической активностью обладает ионизированный кальций. Сдвиг рН плазмы в щелочную сторону уменьшает количество ионизированной формы, а сдвиг в кислую - увеличивает. Депонирование и мобилизация кальция из депо регулируются гормонами щитовидной (кальцитонин) и паращитовидных (ПТГ) желез.
Одна часть кальция выводится почками с мочой, другая экскретируется в кишечник с желчью и панкреатическим секретом. Витамин D усиливает процесс активной реабсорбции кальция в почках, уменьшая его выделение с мочой. Тиазидные диуретики уменьшают экскрецию кальция с мочой.
ПОКАЗАНИЯ
Препараты кальция используют для профилактики и лечения гипокальциемических состояний, а также в комплексной терапии остеопороза. Для этих целей применяют моно- (кальций) и двукомпонентные (кальций + витамин D3 ) ЛС с высоким содержанием кальция.
Среди широко применяемых солей кальция наименьшее количество кальция в 1 г соли содержится в глюконате кальция, наибольшее - в карбонате.
При остро развившейся гипокальциемии, когда уровень общего кальция ниже 1,9 ммоль/л (7,6 мг%), требуются неотложные мероприятия с применением внутривенного введения препаратов кальция: внутривенное медленное (в течение 5 мин) введение 30-50 мл 10% раствора кальция глюконата. В менее экстренных состояниях кальция глюконат разводят в 50 мл 5% раствора декстрозы (глюкозы♠ ) или 0,9% раствора натрия хлорида внутривенно, капельно. Вливание можно повторить при отсутствии положительной динамики в состоянии пациента или сохранении низкого уровня кальция в течение 30 мин, дозы и способы введения меняются в зависимости от состояния пациента (в среднем вводят 15 мг/кг массы тела элементарного кальция за 4-5 ч, увеличивая уровень кальция в крови на 0,5-0,75 ммоль/л.)
В качестве поддерживающей терапии у пациентов с гипокальциемиями любой этиологии, а также при алиментарной недостаточности кальция, для профилактики остеопороза и в комбинации с препаратами для лечения остеопороза используют соли кальция для приема внутрь. Как правило, препараты кальция комбинируют с витамином D. Оптимально применение солей кальция в комбинации с большим содержанием витамина D (из расчета суточного потребления не менее 800 МЕ колекальциферола), в то время как в комбинации с активными метаболитами витамина D можно использовать препараты кальция (500 мг/сут) без витамина D или с его минимальным количеством. Некоторые коммерчески доступные в РФ препараты кальция сведены в табл. 4.30.
| Название препарата | Состав 1 таблетки |
|---|---|
Кальция карбонат + кальция лактоглюконат (Кальций Сандоз Форте♠) (таблетки шипучие) Sandoz, d. d. (Словения), Famar Orleans (Франция) Представительство «Сандоз» (Швейцария) |
Кальция карбонат 1132 мг, кальция лактоглюконат 875 мг, что соответствует 500 мг элементарного кальция |
Кальция карбонат + колекальциферол (Кальций-Д3 Никомед♠) (жевательные таблетки) Nycomed Pharma, AS (Норвегия) Представительство «Такеда» (Япония) |
Кальция карбонат 1250 мг, что соответствует 500 мг элементарного кальция, колекальциферол 200 МЕ |
Кальция карбонат + колекальциферол (Кальций Д3 Классик♠) (жевательные таблетки) Владелец регистрационного удостоверения: Фармацевтическое предприятие «Оболенское», ЗАО (Россия) |
Кальция карбонат 1250 мг, что соответствует 500 мг элементарного кальция, колекальциферол 200 МЕ |
Кальция карбонат + колекальциферол (Кальций-Д3 Никомед Форте♠) (жевательные таблетки) Nycomed Pharma, AS (Норвегия) Представительство «Такеда» (Япония) |
Кальция карбонат 1250 мг, что соответствует 500 мг элементарного кальция, колекальциферол 400 МЕ |
Кальция карбонат + колекальциферол (Компливит кальций Д3 форте♠) (жевательные таблетки) «Фармстандарт-Уфавита», ОАО (Россия) |
Кальция карбонат 1250 мг, что соответствует 500 мг элементарного кальция, колекальциферол 400 МЕ |
Кальция карбонат + колекальциферол (Кальций Д3 Классик♠) (жевательные таблетки) Фармацевтическое предприятие «Оболенское», ЗАО (Россия) |
Кальция карбонат 1250 мг, что соответствует 500 мг элементарного кальция, колекальциферол 400 МЕ |
Кальций-Д3 Актавис♠ Actavis Group hf. (Исландия), производство Индонезия |
Колекальциферол 400 МЕ, кальций 218 мг |
Кальцемин♠ Владелец регистрационного удостоверения: SAGMEL, Inc. (США) Представительство «Байер ХелсКэр АГ» (Германия) |
Кальций (в форме цитрата и карбоната) 250 мг, колекальциферол (вит. D3) - 50 МЕ, медь (в форме оксида) - 500 мкг, цинк (в форме оксида) 2 мг, марганец (в форме сульфата) 500 мкг, бор (в форме натрия бората) 50 мкг |
Кальцемин адванс♠ Владелец регистрационного удостоверения: SAGMEL, Inc. (США) Представительство «Байер ХелсКэр АГ» (Германия) |
Кальций (в форме цитрата и карбоната) 500 мг, колекальциферол (вит. D3) 200 МЕ, магний (в форме оксида) 40 мг, медь (в форме оксида) 1 мг, цинк (в форме оксида) 7,5 мг, марганец (в форме сульфата) 1,8 мг, бор (в форме натрия бората) 250 мкг |
ПРОТИВОПОКАЗАНИЯ
Общие противопоказания к лечению препаратами кальция - гиперкальциемия. Препараты кальция, как правило, противопоказаны при гиперкальциурии, тяжелой почечной недостаточности, нефроуролитиазе, нефрокальцинозе, индивидуальной непереносимости к компонентам препарата. С высокой степенью осторожности препараты кальция применяют при склонности к тромбозам, распространенном атеросклерозе, фибрилляции желудочков, применении сердечных гликозидов, а также при аритмии, ИБС, выраженной гипертензии, в старческом возрасте.
У пациентов с умеренно выраженными нарушениями функции почек, а также при наличии в анамнезе указаний на мочекаменную болезнь необходим регулярный контроль экскреции кальция с мочой с целью коррекции дозы препаратов кальция или отмены лечения ими. Кроме того, больным со склонностью к образованию мочевых камней рекомендуют увеличить потребление жидкости.
ПОБОЧНЫЕ ЭФФЕКТЫ
При приеме внутрь препаратов кальция возможны боли в подложечной области, изжога, нарушения стула.
Подкожное или внутримышечное введение может вызвать некроз тканей и абсцесс. При слишком быстром внутривенном введении возможно появление металлического привкуса во рту, чувства жара, гиперемии лица, брадикардии, гипотонии и даже фибрилляции желудочков.
Чаще побочные эффекты, характерные для внутривенного и внутримышечного путей введения, возникают при использовании хлорида и глюконата кальция.
Повышение содержания кальция в крови (гиперкальциемия) проявляется нарушением стула (запором), жаждой, полиурией, спутанностью сознания.
ВЗАИМОДЕЙСТВИЯ
Принимая во внимание свойства, особенности фармакокинетики и механизма действия препаратов кальция, необходимо учитывать возможность фармакологических взаимодействий с ЛС других групп. Так, в связи с небольшим, но все-таки существующим риском развития гиперкальциемии у больных, принимающих препараты сердечных гликозидов, может увеличиваться возможность появления сердечных аритмий. В таких случаях необходим контроль за уровнем кальция в крови. Тиазидные диуретики уменьшают экскрецию кальция с мочой, поэтому следует учитывать риск развития гиперкальциемии при одновременном назначении их с препаратами кальция.
Препараты кальция нельзя принимать совместно с карбонатами, салицилатами, сульфатами, так как эти соединения образуют нерастворимые или труднорастворимые соли кальция.
Блокаторы Н2 -гистаминовых рецепторов, тормозящие желудочную секрецию, могут оказывать негативное влияние на всасывание кальция.
Препараты кальция могут снижать эффект верапамила и атенолола.
Антипаратиреоидные средства (кальцимиметики)
Для лечения различных форм гиперпаратиреоза и сопутствующей гиперкальциемии был создан агонист кальций-чувствительных рецепторов клеток ОЩЖ - цинакалцет (мимпара♠ ).
ФАРМАКОКИНЕТИКА
Кальций-чувствительные рецепторы, находящиеся на поверхности главных клеток паращитовидных желез, являются основными регуляторами секреции ПТГ. Цинакалцет обладает кальцимиметическим действием, непосредственно снижающим уровень ПТГ, повышая чувствительность рецепторов к внеклеточному кальцию. Снижение ПТГ сопровождается снижением содержания кальция в сыворотке крови.
Снижение уровня ПТГ коррелирует с концентрацией цинакалцета. Вскоре после приема цинакалцета уровень ПТГ начинает понижаться: при этом максимальное снижение происходит примерно через 2-6 ч после приема дозы препарата. Затем концентрация цинакалцета начинает снижаться, а уровень ПТГ увеличивается в течение 12 ч после приема, и далее подавление ПТГ остается примерно на одном и том же уровне до конца суточного интервала при режиме дозирования 1 раз в сутки.
Абсолютная биодоступность цинакалцета при приеме натощак составляла около 20-25%. Прием вместе с пищей увеличивал биодоступность цинакалцета на 50-80%. Подобное повышение концентрации цинакалцета в плазме крови наблюдалось независимо от содержания жира в пище. Связывание с белками плазмы 97%. Метаболизируется преимущественно при участии изоферментов CYP3A4 и CYP1А2 до неактивных метаболитов.
Рекомендуемая начальная доза составляет 30 мг 1 раз в сутки. Затем доза повышается титрованием под контролем содержания ПТГ и кальция в крови. Максимальная доза, в зависимости от показаний и эффективности лечения, может составлять 180-360 мг/сут.
ПОКАЗАНИЯ И ДОКАЗАТЕЛЬНАЯ БАЗА ЭФФЕКТИВНОСТИ
Официальные показания к назначению цинакалцета:
Эффективность цинакалцета у пациентов со вторичным гиперпаратиреозом на фоне хронической почечной недостаточности
В ходе клинических исследований лечение цинакалцетом привело к статистически и клинически значимому улучшению всех метаболических исходов у пациентов со вторичным гиперпаратиреозом, получающих гемодиализ. Статистически значимо (p <0,001) больший процент пациентов достиг снижения интактного ПТГ до уровней, не превышающих 250 пг/мл, а именно 40% пациентов, рандомизированных для получения цинакалцета, и 5%, рандомизированных для получения плацебо, медиана интактного ПТГ, снизилась на 50% в группе, получающей цинакалцет, по сравнению с увеличением на 4% в группе плацебо во время фазы оценки эффективности. Влияние лечения цинакалцетом на концентрацию интактного ПТГ можно ожидать уже через первые 2 нед применения препарата. При применении перитонеального диализа лечение цинакалцетом было также эффективно для снижения уровня интактного ПТГ (≤250 пг/мл) у 38% пациентов.
Значительное снижение значений сывороточного Ca×P, кальция и фосфора, было выявлено упациентов, получавших лечение цинакалцетом, в сравнении с пациентами, получавшими плацебо. После объединенного анализа результатов трех основных исследований (3 фазы) цинакалцет уменьшил значения сывороточного Ca×P на 17% во время фазы оценки эффективности по сравнению с преимущественным отсутствием изменений в группе плацебо. Снижение сывороточного Ca×P было результатом снижения как сывороточного кальция (7%), так и сывороточного фосфора (10%). Важно отметить, что наибольшая эффективность была показана у пациентов с тяжелым вторичным гиперпаратиреозом (>800 пг/мл), которым была рекомендована паратиреоидэктомия, у пациентов, которым были противопоказаны стеролы витамина D в связи с повышенным уровнем кальция и/или фосфора, и у пациентов, не получающих витамин D. Цинакалцет снижал уровни интактного ПТГ и Ca×P независимо от того, имели ли пациенты снижение, отсутствие изменений или увеличение данных показателей при сопутствующей терапии витамином D во время исследования. К окончанию первого года лечения средние значения интактного ПТГ снижались на 44% в группе, получающей цина-калцет, по сравнению с 14%-ным увеличением в группе плацебо. Снижение Ca×P при использовании цинакалцета также сохранялось в течение более чем 12 мес. К окончанию исследования средние значения Ca×P были снижены на 9% от исходного уровня в группе, получающей цинакалцет, по сравнению со снижением на 1% в группе, получающей плацебо.
При вторичном гиперпаратиреозе на додиализной стадии ХПН цинакалцет снизил концентрации интактного ПТГ по сравнению с исходным значением в среднем на 43-59% во время фазы оценки эффективности по сравнению со средним увеличением на 1-6% в группе, получающей плацебо. К тому же 5690 пациентов, получающих лечение цинакалцетом, имели ≥30% снижение от исходного уровня ПТГ во время фазы оценки эффективности по сравнению с 10-28% пациентов, получающими плацебо (значения «р» варьировали от <0,001 до 0,006).
Эффективность при карциноме паращитовидных желез и первичном гиперпаратиреозе
В ходе основного исследования 46 пациентов (29 пациентов с диагнозом «карцинома паращитовидных желез» и 17 - с ПГПТ, у которых паратиреоидэктомия не дала результатов или противопоказана) получали цинакалцет до 3 лет (в среднем 328 дней для пациентов с карциномой паращитовидной железы и 347 дней для пациентов с ПГПТ).
У пациентов с карциномой паращитовидных желез средняя концентрация кальция снижалась с 14,1 до 12,4 мг/дл (3,5 ммоль/л - 3,1 ммоль/л), в то время как у пациентов с ПГПТ концентрация кальция в сыворотке крови снижалась с 12,7 до 10,4 мг/дл (3,2 ммоль/л - 2,6 ммоль/л). У 18 пациентов из 29 (62%) с карциномой паращитовидных желез и 15 пациентов из 17 (88%) с ПГПТ достигнуто снижение концентрации кальция в сыворотке на ≥1 мг/дл ( ≥0,25 ммоль/л).
ПРОТИВОПОКАЗАНИЯ И ОГРАНИЧЕНИЯ ПРИ ПРИМЕНЕНИИ ЦИНАКАЛЦЕТА
Противопоказания: детский и подростковый возраст до 18 лет; повышенная чувствительность к компонентам препарата.
Цинакалцет не следует применять при концентрации кальция в сыворотке крови (с поправкой на альбумин) ниже минимального предела нормального диапазона. Поскольку цинакалцет понижает концентрацию кальция в сыворотке крови, необходимо проводить тщательный мониторинг пациентов в отношении развития гипокальциемии.
В случае гипокальциемии для повышения уровня кальция в сыворотке крови можно использовать кальцийсодержащие препараты, витамин D и/или провести коррекцию концентрации кальция в растворе при диализе. При устойчивой гипокальциемии следует снизить дозу или прекратить применение цинакалцета. Потенциальными признаками развития гипокальциемии могут быть парестезии, миалгии, судороги, тетания.
Цинакалцет не показан пациентам с ХБП, не находящимся на диализе, в связи с возрастанием риска развития гипокальциемии (концентрация сывороточного кальция <8,4 мг/дл или <2,1 ммоль/л) по сравнению с пациентами, находящимися на диализе, что может быть обусловлено более низким начальным уровнем кальция и/или наличием остаточной функции почек.
С осторожностью и под тщательным контролем функции печени следует применять цинакалцет у пациентов с печеночной недостаточностью средней и тяжелой степени (по шкале Чайлда-Пью), так как в таких случаях концентрация цинакалцета в плазме крови может быть в 2-4 раза выше.
При хроническом подавлении концентрации ПТГ ниже концентрации, составляющей приблизительно 1,5% от верхней границы нормы по результатам анализа интактного ПТГ, может развиться адинамическая болезнь кости. Если концентрация ПТГ снижается ниже рекомендуемого диапазона, следует уменьшить дозу цинакалцета и/или витамина D или прекратить терапию.
НЕЖЕЛАТЕЛЬНЫЕ ЯВЛЕНИЯ
Очень часто тошнота, рвота; часто анорексия; иногда диспепсия, диарея; часто головокружение, парестезии; иногда судороги; часто миалгия; часто снижение уровня тестостерона; часто - сыпь; иногда - реакции гиперчувствительности; у пациентов с сердечной недостаточностью регистрировались отдельные идиосинкразические случаи снижения АД и/или ухудшения течения сердечной недостаточности; часто астения, гипокальциемия.
Некоторые побочные реакции цинакалцета могут влиять на способность к управлению автотранспортом или работе с механизмами.
ЛЕКАРСТВЕННОЕ ВЗАИМОДЕЙСТВИЕ
Цинакалцет частично метаболизируется изоферментом CYP3A4. Одновременный прием кетоконазола (сильный ингибитор CYP3A4) в дозе 200 мг 2 раза в сутки приводил к повышению концентрации цинакалцета в плазме примерно в 2 раза. При необходимости одновременного приема мощных ингибиторов (например, кетоконазола, итраконазола, телитромицина, вориконазола, ритонавира) или индукторов CYP3A4 (например, рифампицина) может потребоваться коррекция дозы цинакалцета.
В экспериментальных исследованиях in vitro показано, что цинакалцет частично метаболизируется при участии изофермента CYP1A2. Курение стимулирует активность CYP1A2. Клиренс цинакалцета на 36-38% выше у курящих, чем у некурящих. Влияние ингибиторов CYP1A2 (флувоксамина, ципрофлоксацина) на концентрацию цинакалцета в плазме не изучался. Может потребоваться коррекция дозы, если во время терапии препаратом пациент начинает/прекращает курение или начинает/прекращает одновременный прием мощных ингибиторов CYP1A2.
Цинакалцет является мощным ингибитором CYP2D6. Сочетанное применение цинакалцета и препаратов с узким терапевтическим диапазоном и/или вариабельной фармакокинетикой, метаболизирующихся изоферментом CYP2D6 (например, флекаинид, пропафенон, метопролол, дезипраминρ , нортриптилин, кломипрамин), может потребовать коррекции дозы этих препаратов.
При одновременном приеме цинакалцета в дозе 90 мг 1 раз в сутки с дезипраминомρ (трициклический антидепрессант, метаболизирующийся CYP2D6) в дозе 50 мг повышается уровень экспозиции дезипраминаρ в 3,6 раза у пациентов с активным метаболизмом CYP2D6.
Препараты фосфора и фосфатбиндеры
Фосфор широко распространен в природе и составляет около 0,9% массы Земли. В естественных условиях в чистом виде он не встречается, так как обладает высокой химической активностью и быстро образует химические соединения. Суточное потребление фосфора составляет 1200-1600 мг у взрослого человека. Бóльшая часть фосфора находится в костной ткани, обеспечивая эластичные свойства гидроксиаппатита. Фосфор входит в состав АТФ, ДНК, некоторых аминокислот и, следовательно, необходим для энергетического обмена и сохранения генетической информации. В норме человек получает фосфор с пищей. Больше всего фосфора содержится в сухих дрожжах - 1290 мг на 100 г продукта, на втором месте молочные продукты, затем морепродукты. Лучше всего фосфор (90%) усваивается из молочных продуктов и хуже (20%) из растительных. При этом здоровый человек безопасно может употреблять неограниченное количество фосфора. Обмен фосфора осуществляется при взаимодействии активной формы витамина D (D-гормона), фактора роста фибробластов 23 и опосредованно ПТГ. D-гормон повышает всасывание фосфора в кишечнике, ПТГ повышает высвобождение фосфора из костной ткани при ее резорбции, а фактор роста фибробластов 23, напротив, усиливает выведение фосфора с мочой и переводит D-гормон в неактивную форму, тем самым уменьшая всасывание фосфора в кишечнике.
Низкое содержание фосфора встречается достаточно редко: при фосфопенических формах остеомаляции, а также в случае эктопической секреции опухолью фактора роста фибробластов 23.
Для компенсаций нарушений фосфорного обмена при гипофосфатемии в ряде случаев используются препараты фосфора [внутривенно фосфат натрия или калия при уровне фосфора менее 0,8 нмоль/л в дозах 0,08 ммоль/кг (2,5 мг/кг) до 0,16 ммоль/кг (5 мг/кг) в течение 6 ч или в таблетках 1000-2000 мг (32-64 ммоль/л) фосфора в день (7-10 дней)], активные метаболиты витамина D: альфакальцидол, кальцитриол в высоких дозах.
На этапе клинических исследований находятся моноклональные антитела к фактору роста фибробластов 23.
ВЗАИМОДЕЙСТВИЕ С ДРУГИМИ ВЕЩЕСТВАМИ
При приеме препаратов фосфора следует помнить, что увеличение его количества со съеденной пищей ощутимо затрудняет всасывание магния. Если в суточном рационе кальция будет больше, чем фосфора, то биодоступность фосфора уменьшается. Всасывание фосфора улучшается, если употреблять его совместно с витаминами A, D, F.
ФОСФАТБИНДЕРЫ
При серьезных нарушениях функции почек может наблюдаться гиперфосфатемия.
Пациентам на гемодиализе показано соблюдение гипофосфатной диеты, что не всегда удается. Кроме того, при соблюдении гипофосфатной диеты в связи с ограничением потребления белка наблюдается недостаточное потребление калорий, гипоальбуминемия, что связано с увеличением заболеваемости и смертности у пациентов с ХБП. В связи с этим становится актуальным применение фосфатбиндеров.
Фосфатбиндеры (препараты, связывающие фосфор и понижающие его содержание в крови) бывают нескольких видов:
ПРЕПАРАТЫ, СОДЕРЖАЩИЕ АЛЮМИНИЙ
Эффективными фосфатбиндерами являются препараты, содержащие алюминий, однако существует опасность развития алюминиевой интоксикации с микроцитарной анемией, остеомаляцией и поражением ЦНС. В связи с этим вопрос о назначении данных препаратов остается спорным. В настоящее время их назначают короткими курсами для коррекции уровня фосфора.
ПРЕПАРАТЫ НА ОСНОВЕ КАЛЬЦИЯ
Фосфатбиндеры на основе кальция эффективно снижают уровень фосфора и ПТГ, но при этом увеличивается риск гиперкальциемии и кальцифилаксии, так как 20-30% принятого кальция поступает в кровоток. Самыми популярными из фосфатбиндеров, содержащих соли кальция, являются кальция карбонат и кальция ацетат. Однако терапия кальция карбонатом в больших дозах (5,5- 6,0 г/сут) связана с гиперкальциемией у 20% пациентов. При сравнении ацетата кальция с кальция карбонатом гиперкальциемия встречается реже. Обнаружена прямая связь между дозой назначенного кальция карбоната и ригидностью артериальной стенки, связанной с наличием в ней кальция. В связи с потенциальным риском сердечно-сосудистой кальцификации рекомендуется применение 1-2 г элементного кальция в день. При назначении препаратов витамина D увеличивается риск возникновения внескелетных кальцификаций. Goodman и соавт. показали, что молодые диализные пациенты с кальцификацией коронарных артерий, доказанной по КТ, употребляли в два раза больше кальцийсодержащих фосфатбиндеров, чем пациенты без депозитов кальция.
Необходимо избегать назначения кальция цитрата пациентам с уремией, особенно в случае применения препаратов, содержащих алюминий. Вследствие того что цитрат кальция способствует абсорбции алюминия через кишечник, может развиться алюминиевая интоксикация.
ПРЕПАРАТЫ, НЕ СОДЕРЖАЩИЕ КАЛЬЦИЙ И АЛЮМИНИЙ
С учетом всех осложнений, возникающих при применении фосфатбиндеров, содержащих кальций и алюминий, были созданы препараты, не абсорбирующие кальций и не содержащие алюминий: севеламер и лантана карбонат. Фосфатбиндеры, не содержащие кальций, эффективно понижают уровень фосфора сыворотки, на определенное время могут остановить увеличение уровня ПТГ, но прямо не влияют на уровень ПТГ. Севеламер хорошо переносится пациентами, значительно замедляет прогрессию сосудистой кальцификации, однако по сравнению с препаратами, содержащими кальций, при длительном лечении хуже снижает уровень фосфора у гемодиализных пациентов. Для усиления подавления уровня ПТГ препарат может эффективно использоваться в комбинации с препаратами кальция и витамина D. Благоприятное плейотропное действие на липидный профиль (уменьшает уровень общего холестерина, ЛПНП, не влияет на уровень ЛПВП) может уменьшить сердечно-сосудистый риск, связанный с дислипидемией. Однако при приеме севеламера увеличивается риск развития метаболического ацидоза в связи с уменьшением бикарбоната сыворотки. Плацебоконтролируемое исследование (продолжительность 17 мес), проведенное Collins и соавт. у гемодиализных больных, показало, что у пациентов, принимающих севеламер, риск госпитализации был ниже, чем у пациентов группы контроля.
Необходимо учитывать, что севеломер снижает биодоступность ципрофлоксацина на 50%.
Препараты для лечения остеопороза
Препараты для лечения остеопороза вмешиваются в костное ремоделирование и опосредованно могут влиять на фосфорно-кальциевый обмен. В настоящее время для лечения остеопороза в РФ применяются следующие ЛС: терипаратид, деносумаб, азотсодержащие бисфосфонаты [алендроновая кислота (алендронат♠ ), ризедронат, ибандроновая кислота, золедроновая кислота], стронция ранелат.
В зависимости от преимущественного влияния на процессы костного ремоделирования выделяют препараты с анаболическим действием на кость (преимущественно стимулируют костеобразование) - терипаратид (форстео♠ ) или антирезорбтивным эффектом (преимущественно подавляют костное разрушение) - деносумаб (пролиа♠ ), бисфосфонаты. Селективные модуляторы эстрогеновых рецепторов (ралоксифен, базедоксифенρ , лазофоксифенρ ) также обладают антирезорбтивным и рядом плейотропных эффектов, однако в России отсутствуют.
ПРЕПАРАТЫ ПАРАТИРЕОИДНОГО ГОРМОНА
Основной точкой приложения для реализации эффекта терипаратида является остеобласт. На фоне лечения терипаратидом костный обмен повышается с преобладанием костеобразования, т.е. сначала повышаются маркеры костеобразования (на 150-200% N-концевой пропептид коллагена первого типа) и только потом (с формированием терапевтического окна для анаболического эффекта) - маркеры костного разрушения (на 58% для N-телопептида в моче). Более того, исследования биопсий костной ткани человека позволили продемонстрировать, что наряду с общим повышением костного обмена наблюдаются процессы костеобразования, соответствующие моделированию, т.е. формированию новой молодой костной ткани, как это происходит у ребенка и подростка во время набора пика костной массы.
Механизмы повышения костеобразования в ответ на введение интермитирующих доз ПТГ являются предметом пристального изучения. Наиболее очевидным кажется взаимодействие ПТГ/ПТГ-связанный пептид. Через рецептор к ПТГ увеличивается экспрессия гена Runx2 , повышается экспрессия других генов, способствующих дифференцировке остеобластов, в результате чего повышается остеобластогенез и выживаемость остеобластов. Однако по мере накопления знаний становится очевидной вовлеченность нескольких сигнальных путей. Есть доказательства того, что анаболическое действие ПТГ реализуется через активацию канонического Wnt-сигнала. ПТГ ингибирует экспрессию склеростина и диккопфа-1 (Dkk-1) - основных ингибиторов Wnt-сигнала. Активация Wnt-сигнального пути вносит свой вклад в анаболический эффект терипаратида в трабекулярной костной ткани и влияет на повышение периостального костеобразования.
В основное исследование эффективности терипаратида - Fracture Prevention Trial (III фаза) - было включено 1637 женщин в постменопаузе. Женщины, включенные в исследование, должны были иметь как минимум один низкотравматичный перелом средней степени (снижение высоты тела позвонка на 26-40%) или два легких (снижение высоты тел позвонков на 20-25%) низкотравматичных перелома тел позвонков. Для тех пациентов, у кого было меньше двух переломов позвонков средней тяжести, оговаривалась необходимость снижения МПК как минимум на 1 стандартное отклонение (Т-критерий ≤-1,0), т.е. терипаратид стал фактически единственным препаратом, в критериях включения которого не требовалось снижения МПК до -2,5 по Т-критерию хотя бы в одном из отделов.
Включенные пациенты (средний возраст 69 лет) были рандомизированы на группы, получавшие плацебо (544 человека), 20 мкг (541 человек) и 40 мкг (552 человека) терипаратида в сутки в виде ежедневных подкожных инъекций. Все пациенты принимали препараты кальция (1000 мг/сут) и витамина D (400- 1200 ЕД/сут). Продолжительность лечения составила в среднем 18 мес.
Риск возникновения новых переломов тел позвонков у пациентов, получавших 20 мкг терипаратида, был снижен на 65% по сравнению с плацебо. Риск внепозвоночных переломов был снижен на 53% в группе 20 мкг по сравнению с плацебо. Лечение терипаратидом позволило уменьшить риск тяжелых переломов позвонков на 90%. Риск переломов позвонков статистически значимо снижался независимо от наличия предшествующих переломов и степени их тяжести. Эффективность терапии не зависела от возраста, снижения МПК и количества предшествующих переломов. Последующий анализ рентгенологический снимков позвоночника пациентов, включенных в это исследование, проводился с анализом количественным методом (снижение высоты тела позвонка на 20% и более) и полуколичественным методом при визуальной оценке. При такой комплексной оценке было получено снижение риска новых переломов позвонков на 84% (относительный риск = 0,16p <0,001), множественных переломов позвонков - на 94% (относительный риск = 0,06p <0,001) у пациентов, получавших терипаратид, по сравнению с плацебо.
Анаболическое действие терипаратида отражалось в дозозависимом увеличении преимущественно маркеров костеобразования, а также в меньшей степени маркеров костной резорбции. По результатам исследования было выявлено повышение МПК позвонков на 10-14%, повышение МПК шейки бедра на 3-5% за 18 мес терапии.
Положительная динамика МПК (минимум +3%) наблюдалась у 91% женщин, получавших терипаратид 20 мкг. Эффективность препарата в отношении снижения риска переломов не зависела от исходного уровня маркеров костного обмена.
Терипаратид выпускается в стеклянном картридже, который укреплен в одноразовую ручку, рассчитанную для введения 28 доз. Подкожные иньекции в дозе 20 мкг производятся самостоятельно пациентом 1 раз в сутки, ежедневно. Хранить препарат необходимо в холодильнике, однако в течение несколько часов возможно пребывание при комнатной температуре.
Особенностью лечения терипаратидом является проведение 1 курса лечения в течение жизни, длительностью до 24 мес максимально.
На основании полученных в исследованиях данных предполагается перед назначением препарата проводить следующее обследование пациентов: общий кальций сыворотки, общая ЩФ сыворотки, 25-гидроксивитамин D ПТГ, клиренс креатинина.
Безопасность применения терипаратида
Наиболее частыми побочными эффектами (менее 10% испытуемых) были головокружение (ортостатическая гипотензия, которая обычно не требовала прерывать лечение, случалась при введении первых доз терипаратида особенно при выполнении инъекции лежа) и судороги в ногах. Тошнота и головная боль носили дозозависимый характер и достоверно чаще встречались у пациентов, получавших 40 мкг терипаратида. Умеренная транзиторная гиперкальциемия (менее 2,8 ммоль/л) была зарегистрирована у 2% группы контроля после иньекции, 11% - у пациентов, получавших 20 мкг терипаратида. Транзиторная гиперкальциемия встречалась обычно в первые 6 мес лечения терипаратидом. Увеличение уровня кальция наблюдалось через 4-6 ч после инъекции и полностью нормализовывалось через 24 ч. Увеличение кальциурии отличалось недостоверно от группы контроля и не ассоциировалось с увеличением риска мочекаменной болезни. Аллергические реакции были редки. Антитела к терипаратиду были обнаружены у 3-8% женщин, но со временем их содержание уменьшилось, и они не оказали никакого эффекта на МПК или уровень кальция.
Применение терипаратида у пациентов с умеренным снижением СКФ (30- 49 мл/мин) приводило к увеличению мочевой кислоты в сыворотке крови, но это не сочеталось с увеличением риска развития подагры, артралгией или камнеобразованием в почках.
Противопоказания для назначения терипаратида
Гиперкальциемия, болезнь Педжета, необъяснимое повышение ЩФ, остеогенная саркома, незакрытые зоны роста, облучение скелета в анамнезе, беременность или кормление грудью, рак кости или метастазы рака в кости, аллергическая реакция к терипаратиду или компонентам растворителя.
Взаимодействие терипаратида с другими веществами
Клинически значимого взаимодействия с гидрохлоротиазидом (гидрохлортиазидом♠ ), фуросемидом, дигоксином, атенололом, а также с препаратами с замедленным высвобождением - дилтиаземом, нифедипином, фелодипином, нисолдипиномρ - не отмечалось.
Совместное назначение терипаратида с ралоксифеном или гормонозаместительной терапией не влияет на содержание кальция в сыворотке крови и в моче.
АНТИТЕЛА К РЕЦЕПТОРУ АКТИВАТОРУ ЯДЕРНОГО ФАКТОРА Κ-B
Деносумаб - человеческое моноклональное антитело к RANKL. Препарат обладает высоким сродством к RANKL и высокой специфичностью, т.е. не связывается с родственными молекулами (фактором некроза опухоли-α и -β, апоптоз-вызывающий лиганд, зависимый от фактора некроза опухоли, СD40L).
Антитело к RANKL имеет значительно больший период жизни и соответственно реже дозируется, в отличие от остеопротегерина. В отличие от других антирезорбтивных препаратов - бисфософонатов, деносумаб уменьшает образование остеокластов, а не нарушает функцию зрелых клеток. Кроме того, будучи биологическим препаратом, деносумаб не накапливается в костной ткани и не оказывает отсроченного влияния с полным обратным развитием эффекта после отмены лечения.
Эффективность деносумаба для предупреждения низкотравматичных переломов у женщин в постменопаузе исследовалась в многоцентровом, рандомизированном, двойном слепом плацебоконтролируемом исследовании с участием 7868 женщин 60-90 лет (средний возраст 72 года) с постменопаузальным остеопорозом (Т-критерий в поясничных позвонках или в целом в бедре от -2,5 до -4,0; 23,8% пациенток имели предшествующие переломы позвонков). Пациенты получали лечение препаратом деносумаб 60 мг подкожно или плацебо в течение 36 мес, все женщины дополнительно получали кальций и витамин D.
За три года лечения деносумабом наблюдалось снижение риска морфометрических переломов позвонков на 68% p <0,001 (2,3% в группе деносумаба по сравнению с 7,2% в группе плацебо); переломов бедра на 40% (p=0,04; 0,7% в группе деносумаба по сравнению с 1,2% в группе плацебо) и на 20% снизился риск внепозвоночных переломов (p=0,01; 6,5% в группе деносумаба по сравнению с 8,0% плацебо).
К настоящему времени эффективность лечения деносумабом доказана при восьмилетнем лечении, МПК и прочность кости повышаются, а риск переломов снижается в костях как с преимущественным содержанием трабекулярной, так и кортикальной костной ткани. Препарат эффективно повышает МПК во всех отделах скелета после терапии терипаратидом. При прямых сравнительных исследованиях деносумаба и бисфосфонатов для приема внутрь деносумаб статистически значимо больше повышал МПК во всех отделах скелета, в том числе в лучевой кости. Благодаря системному распределению деносумаб оказывает лучшие эффекты для прибавки МПК в кортикальной кости по сравнению с бисфосфонатами. Кроме того, при комбинации терипаратида и деносумаба удалось добиться лучшей прибавки МПК по сравнению с применением только терипаратида.
Деносумаб выпускается в оригинальной упаковке-шприце с предварительно набранным раствором 1 мл - 60 мг для подкожных инъекций, 1 раз в 6 мес. Хранится препарат при температуре 3-6 °С. Чтобы избежать дискомфорта в месте введения, следует согреть paствор до комнатной температуры (до 25 °С) перед инъекцией, а затем медленно ввести все содержимое предварительно заполненного шприца. После инъекции шприц выбрасывается.
Безопасность применения деносумаба
В ходе применения деносумаба для лечения постменопаузального остеопороза были выявлены следующие нежелательные явления:
-
иногда (с частотой от 1 на 1000 пациентов до 1 на 100 пациентов) экзема (включая дерматиты аллергические, атопические, контактные), целлюлит, дивертикулит, отогенные инфекции;
-
редко (с частотой от 1 на 1000 пациентов до 1 на 10 000 и <1 из 1000) остеонекроз челюсти, гипокальциемия (в основном у пациентов с почечной недостаточностью);
-
очень редко (<1 на 10 000 пациентов, получающих лечение) атипичный перелом бедренной кости (отчет о безопасности программы клинических исследований по остеопорозу).
Деносумаб не рекомендован к применению у детей, так как эффективность и безопасность данного препарата не изучались в этой возрастной группе.
Основываясь на имеющихся данных об эффективности и безопасности препарата, у пациентов пожилого возраста и у пациентов с почечной недостаточностью не требуется коррекции режима дозирования препарата. У пациентов с тяжелой почечной недостаточностью (СКФ <30 мл/мин) или находящихся на диализе существует большой риск развития гипокальциемии. Эффективность и безопасность применения препарата при печеночной недостаточности не изучались.
Противопоказания для назначения деносумаба
Гипокальциемия, детский и подростковый возраст до 18 лет, наследственная непереносимость фруктозы, беременность, период лактации, повышенная чувствительность к деносумабу или любому из компонентов препарата.
Взаимодействия
Деносумаб не оказывает влияния на фармакокинетические показатели препаратов, метаболизирующихся системой CYP3A4. Другие взаимодействия не описаны.
БИСФОСФОНАТЫ
Свое название эта группа ЛС получила благодаря существованию в химической структуре атома углерода, связанного с двумя атомами фосфора (негидролизируемая P - C - P-связь). По своей структуре бисфосфонаты схожи с эндогенным пирофосфатом и были созданы по его подобию первоначально для лечения эктопической кальцификации у пациентов с кальцифилаксией. Неорганический пирофосфат в норме содержится у здоровых людей (средняя концентрация в плазме 3,5±0,11 мкмоль/л с референтным интервалом 1,19-5,65 мкмоль/л) и препятству-ет выпадению кальция в осадок в крови и моче. В костной ткани эндогенный пирофосфат разрушается ЩФ, и, таким образом, происходит минерализация кости. Бисфосфонаты способны активно связываться с кристаллами гидроксиапатита костной ткани, но при этом последнее поколение препаратов - аминобисфосфонаты [алендроновая кислота (Алендронаталендронат♠ ), ризедронат, ибандронат натрия, золедроновая кислота (Золедронатзоледронат-тева♠ )] - не нарушают минерализации кости.
Механизм действия и фармакологические эффекты
Механизм действия бисфосфонатов основан на том, что они тормозят резорбтивную активность остеокластов. Бисфосфонаты первого поколения (этидронат, клодронат и тилудронат) метаболизируются с образованием соединений - аналогов АТФ. Эти соединения накапливаются в остеокластах и нарушают их функцию. Аминобисфосфонаты действуют иначе: они ингибируют фарнезилпирофосфатазу и другие этапы метаболизма мевалоната, что приводит к снижению резорбтивной активности остеокласта, нарушению его цитоскелета и стимулирует апоптоз остеокласта. Присутствие атома азота в боковой цепи молекулы бисфосфоната существенно повышает антирезорбтивные способности этих ЛС.
Бисфосфонаты плохо всасываются в ЖКТ, абсорбция при приеме пищи уменьшается, поэтому таблетированные бисфосфонаты следует принимать натощак минимум за 30 мин до еды, запивая питьевой водой (1 стакан). Достаточно часто используют внутривенные бисфосфонаты, которые не оказывают влияния на верхние отделы ЖКТ. Выведение бисфосфонатов осуществляется в две фазы: первая - быстрая (обычно несколько часов) и вторая - медленная (несколько лет). Вторая фаза выведения связана с медленным высвобождением бисфосфонатов из костей. Бисфосфонаты не метаболизируются, поэтому выводятся с мочой практически в неизмененном виде.
Крупные плацебо-контролируемые исследования показали, что все современные бисфосфонаты эффективны для предупреждения переломов тел позвонков, переломов бедра и других внепозвоночных переломов, повышения МПК и снижения маркеров костного разрушения у женщин в постменопаузе.
Средняя продолжительность лечения - 3-5 лет, есть данные о 10-летнем непрерывном применении алендроновой кислоты.
Бисфосфонаты длительно остаются в костной ткани, их период полувыведения может достигать 10 лет, поэтому возможно интермитирующее применение этих препаратов с перерывами после трех, пяти лет лечения.
Помимо постменопаузального остеопороза, бисфосфонаты с успехом применяют при ювенильном, глюкокортикоидном и иммобилизационном остеопорозе, а также при остеопорозе у мужчин.
Бисфосфонаты считают ЛС выбора для лечения болезни Педжета костей (деформирующий остеит), а также гиперкальциемии опухолевого генеза. Как правило, при болезни Педжета применяют алендроновую кислоту (алендронат♠ ) и золедроновую кислоту (золедронат-тева♠ ). При гиперкальциемии опухолевого генеза обычно рекомендуют ибандронат натрия, памидроновую кислоту и золедроновую кислоту (золедронат-тева♠ ) в больших дозах. Бисфосфонаты могут использовать и при гиперкальциемии иной этиологии (например, при гиперпаратиреозе).
Побочные эффекты
Бисфосфонаты в большинстве случаев хорошо переносят, побочные эффекты редко заставляют отказаться от терапии. Основной проблемой при приеме внутрь считают нарушения со стороны ЖКТ: прежде всего боли в эпигастральной области (за счет эзофагита, обострения гастрита или язвенной болезни), реже запор, диарею, метеоризм и дисфагию.
При внутривенном введении бисфосфонатов возможно повышение температуры тела, иногда гриппоподобный синдром (лихорадка, озноб, боли в костях и мышцах). В большинстве случаев специфического лечения при подобном состоянии не требуется, поскольку симптомы исчезают в течение первых суток.
Могут возникать различные дерматологические реакции (сыпь, эритема). Очень редко развиваются побочные эффекты со стороны органа зрения (боль, нарушение зрения, конъюнктивиты, увеиты, склериты).
В постмаркетинговых исследованиях обсуждается связь применения бисфосфонатов с развитием остеонекроза челюсти и атипичных переломов. Однако эти нежелательные явления очень редки.
Противопоказания
Бисфосфонаты противопоказаны у пациентов с гипокальциемией, выраженными нарушениями функции почек (СКФ менее 35 мл/мин), повышенной чувствительностью к бисфосфонатам, в период беременности и лактации. Для таблетированных форм бисфосфонатов противопоказанием являются активные эрозивно-язвенные процессы в верхних отделах ЖКТ.
Взаимодействия
После приема таблетированных форм бисфосфоната рекомендуют в течение 30 мин воздерживаться от приема других ЛС, особенно содержащих многовалентные катионы (препараты кальция, антациды и проч.), чтобы избежать нарушения абсорбции бисфосфонатов.
Прием ацетилсалициловой кислоты и других НПВС на фоне терапии бисфосфонатами может сопровождаться увеличением риска развития побочных эффектов со стороны ЖКТ.
Блокаторы H2 -гистаминовых рецепторов (в частности, ранитидин) за счет увеличения рН желудочного сока могут повышать биодоступность бисфосфонатов [алендроновая кислота (алендронат♠ )].
Рекомендуется соблюдать осторожность при назначении бисфосфонатов с аминогликозидами, поскольку ЛС обеих групп понижают уровень кальция в сыворотке на длительное время.
ПРЕПАРАТ СТРОНЦИЯ
Стронция ранелат - соль стронция - выпускается в саше по 2 г стронция для ежедневного приема. Оказывает действие, подобное высоким дозам кальция, встраивается в кристаллическую решетку гидроксиаппатита, что делает кость тяжелее и плотнее. В рандомизированных клинических исследованиях доказана эффективность стронция ранелата для снижения риска переломов позвонков за 3 года на 41% и риска внепозвоночных переломов на 16%. При вторичном анализе данных в субпопуляции пациентов старше 74 лет с выраженным остеопорозом в шейке бедра (Т-критерий ниже или равен -3) препарат снижал риск переломов бедра на 36% (p=0,046).
По всей видимости, атомы стронция по аналогии с кальцием вмешиваются и в другие важные биологические сигнальные пути. По данным метаанализов рандомизированных клинических исследований (n =7400) и постмаркетинговых наблюдений выявлено, что риски от применения стронция ранелата для лечения остеопороза могут превосходить потенциальную пользу у пациентов с патологией сердечно-сосудистой системы и склонностью к тромбозам: так, препарат повышает риск инфаркта миокарда на 62%, тромбозов на 50%. В настоящее время применение стронция ранелата ограниченно. Стронция ранелат может быть рекомендован только пациентам с тяжелым остеопорозом, у которых ни один другой препарат не подходит, при условии, что на момент назначения стронция ранелата у пациентов нет кардиоваскулярных заболеваний, в том числе заболеваний периферических артерий, рисков тромбоза и тромбоэмболии, плохо контролируемой гипертензии. Лечение должно проводиться при постоянном мониторинге состояния кожи и кардиоваскулярных рисков.
Дозы всех препаратов для лечения остеопороза и показания для их применения сведены в табл. 4.31.
ПРЕПАРАТЫ НА ЭТАПЕ РАЗРАБОТОК
Заканчиваются клинические исследования нескольких препаратов для лечения остеопороза: новый антирезорбтивный препарат - оданокатиб - ингибитор катепсина К; новые анаболические препараты: ромазосумаб и блазосумаб - моноклональные антитела к склеростину.
Проходят клинические исследования применения моноклонального антитела к фактору роста фибробластов 23 - препарат для повышения уровня фосфора в сыворотке крови у пациентов с опухолью, секретирующей фактор роста фибробластов 23 или фосфопеническими формами остеомаляций, связанными с генетическими нарушениями в метаболизме фактора роста фибробластов 23.
Заканчиваются клинические исследования препарата заместительной терапии асфотаза альфаρ для лечения гипофосфатазии.
Препарат |
Доза и способ введения |
Показание |
|||
Пocтмeнoпayзaльный остеопороз |
Остеопороз у мужчин |
Γлюкoкopтикoидный остеопороз |
Профилактика ocтeoпopoзa |
||
Aлeндpoнoвaя кислота (aлeндpoнaт♠) |
Таблетка 70 мг 1 раз в неделю |
+ |
+ |
+ |
+ (35 мг 1 раз в неделю) |
Ибaндpoнaт натрия |
Таблетка 150 мг 1 раз в мес. Раствор для внутривенной инфyзии 3,0 мл 1 раз в 3 мес внутривенно |
+ |
|||
Pизeдpoнaт |
35 мг 1 раз в неделю |
+ |
+ (у оригинального препарата aктoнeля♠) |
+ (у оригинального препарата aктoнeля♠) |
|
Зoлeдpoнoвaя кислота (зoлeдpoнaт-тeвa♠) |
Раствор 5,0 мг в 100 мл 1 раз в год внутривенно |
+ |
+ |
+ |
+ (1 инфyзия в 2 года) |
Дeнocyмaб |
Раствор 60 мг в 1 мл подкожно 1 раз в 6 мес |
+ |
+ |
||
Терипаратид |
20 мкг подкожно ежедневно (шприц-ручка 250 мкг в 1 мл) |
+ |
+ |
+ |
|
Стронция paнeлaт |
Порошок (саше) по 2 г, ежедневно через 2 ч после последнего приема пищи |
+ |
+ |
||
СПИСОК ЛИТЕРАТУРЫ
-
Белая Ж.Е. Рациональный выбор фармакотерапии постменопаузального остеопороза. Эффективность и безопасность бонвивы: обзор за восемь лет применения // Остеопороз и остеопатии. - 2013. - № 2. - С. 22-30.
-
Белая Ж.Е., Рожинская Л.Я. Бисфосфонаты в терапии постменопаузального остеопороза // Доктор. Ру. - 2010. - Т. 58. - № 7. - С. 29-38.
-
Белая Ж.Е., Рожинская Л.Я. Витамин D в терапии остеопороза: его роль в комбинации с препаратами для лечения остеопороза, внескелетные эффекты // Эффективная фармакотерапия. - 2013. - № 2. - С. 14-29.
-
Белая Ж.Е., Рожинская Л.Я. Бисфосфонаты в терапии постменопаузального остеопороза // Доктор. Ру. - 2010. - Т. 58. - № 7. - С. 29-38.
-
Белая Ж.Е., Рожинская Л.Я. Анаболическая терапия остеопороза. Терипаратид: эффективность, безопасность и область применения // Остеопороз и остеопатии. - 2013. - № 2. - С. 32-40.
-
Егшатян Л.В., Рожинская Л.Я., Кузнецов Н.С. Медикаментозные методы лечения вторичного гиперпаратиреоза у пациентов с хронической болезнью почек // Медицинский совет. - 2010. - № 1-2.
-
Михайлова Н.А. Селективный активатор витамин D-рецепторов парикальцитол и его место в лечении хронической болезни почек. Обзор литературы // Лечащий врач. - 2011.
-
Письмо Федеральной службы по надзору в сфере здравоохранения от 26 июля 2013 г. №16И-824/13 «О новых данных по безопасности лекарственного препарата Бивалос». http://www.garant.ru/products/ipo/prime/doc/70322734/#ixzz3L7zAbNI1.
-
Рожинская Л.Я., Ростомян Л.Г., Егшатян Л.М. Новые возможности лечения гиперпаратиреоза модуляторами кальций-чувствительного рецептора (кальцимимети-ками) // Остеопороз и остеопатии. - 2008. - Вып. 2.
-
Block G.A., De Francisco A.L. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysys // N. Engl.J. Med. - 2004. - Vol. 350. - P. 1516-1525.
-
Chertow et al. Cinacalcet hydrochloride (Sensipar) in hemodialysis patients on active vitamin D derivatives with controlled PTH and elevated calcium x phosphate // Clinical Journal of the American Society of Nephrology. - 2006.
-
Dempster D.W., Zhou H., Recker R.R. et al. Skeletal histomorphometry in subjects on teriparatide or zoledronic acid therapy (SHOTZ) study: a randomized controlled trial // J. Clin. Endocrinol. Metabolism. - 2012. - Vol. 97. - P. 2799-2808.
-
European Medicines Agency: Recommendation to restrict the use of Protelos/Osseor (strontium ranelate) 25 April 2013 EMA/258269/2013.
-
Gallagher J.C., Genant H.K., Crans G.G. et al. Teriparatide reduces the fracture risk associated with increasing number and severity of osteoporotic fractures // J. Clinical Endocrinology Metabolism. - 2005. - Vol. 90. - P. 1583-1587.
-
Holick M.F., Binkley N.C., Bischoff-Ferrari H.A. et al. Evaluation, treatment, and prevention of Vitamin D deficiency: an Endocrine Society Clinical Practice Guideline // JCEM. - 2011. - Vol. 96. - P. 1911-1930.
-
Lindberg J.S. et al. Cinacalcet, an oral calcimimetic agent for the treatment of secondary hyperparathyroidism in hemodialysis and peritoneal dialysis: a randomized, double-blind, multicenter study // Journal of the American Society of Nephrology. - 2005.
-
Lindberg J.S. et al. The calcimimetic AMG073 reduces parathyroid hormone and calciumх phosphorus in secondary hyperparathyroidism // Kidney Int. - 2003. - Vol. 63. - P. 248-254.
-
Nakane M., Ma J., Rose A.E. et al. Differential effects of vitamin D analogs on calcium transport // J. Steroid. Biochem. Mol. Biol. - 2007. - Vol. 103. - P. 84-89.
-
National Kidney Foundation 2003 K/DOQI Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease // Am.J. Kidney Dis. - 2003. - Vol. 42 (supp. l). - P. 1-202. (Перевод на рус. яз.: Нефрология и диализ. ISSN 1680-4422, 2008.)
-
Neer R.M., Arnaud C.D., Zanchetta J.R. et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis // England J. Medicine. - 2001. - Vol. 35. - P. 1434-1441.
-
Saag K.G., Shane E., Boonen S. et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis // New England J. Medicine. - 2007. - Vol. 357. - P. 2028-2039.
-
Sterz R., Frye С., Khan S. et al. Paricalcitol treatment in CKD patients with secondary hyperparathyroidism associated with better health outcomes when compared with no vitamin D receptor (VdR) activator treatment / National Kidney Foundation, Spring Clinical Meeting Orlando, Florida, April 14-16, 2010 // Abstract#Poster. - 2010. - N 257.
-
Sterrett J.R. et al. Cinacalcet HCl (SensiparAMimparaA is an effective chronic therapy for hemodialysis patients with secondary hyperparathyroidism // Clin. Nephrol. - 2007. - Vol. 68. - P. 10-17.
-
Teng M., Wolf M., Lowrie E. et al. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy // N. Engl. J. Med. - 2003. - Vol. 349. - P. 446-456.
ПРЕПАРАТЫ, СНИЖАЮЩИЕ МАССУ ТЕЛА
Гурова О.Ю., Колода Д.Е., Мазурина Н.В.
Изменение пищевого рациона и снижение физической активности приводят к ежегодному увеличению числа людей, страдающих ожирением. При этом лечение данного заболевания - крайне непростой процесс, направленный не только на снижение массы тела, но прежде всего на удержение достигнутого результата. При этом фармакотерапия - это лишь вспомогательное средство. Основой же лечения является изменение образа жизни, подразумевающее расширение физической активности и соблюдение рекомендаций по питанию.
КЛАССИФИКАЦИЯ
Основные группы препаратов, снижающих массу тела:
Длительное время в клинической практике отсутствовали эффективные и безопасные ЛС для лечения ожирения. ЛС из ранее применявшихся препаратов группы симпатомиметиков не разрешены для лечения ожирения. Некоторые препараты этой группы (фенфлурамин, дексфенфлурамин, эфедрин) включены в список сильнодействующих веществ.
Использование антидепрессантов в лечении ожирения весьма ограниченно. Тем не менее указанные препараты (особенно флуоксетин) рекомендуют для лечения пациентов с ожирением и депрессивными расстройствами, сопровождаемыми нарушениями пищевого поведения. По такому же принципу для лечения ожирения назначают противоэпилептические препараты.
Больным СД 2-го типа с ожирением или избыточной массой тела показаны препараты, снижающие уровень глюкозы в плазме крови, дополнительно уменьшающие или стабилизирующие массу тела.
Римонабантρ - первый представитель класса блокаторов каннабиноидных рецепторов I типа - был разработан для лечения ожирения и прошел клинические испытания. Римонабантρ не был разрешен для использования в широкой клинической практике (в том числе не получил одобрения Управления по контролю за пищевыми продуктами и ЛС США - Food and Drug Administration) из-за рисков, связанных с его применением (в частности, возникновения суицидальных мыслей). Наиболее значимыми побочными эффектами римонабантаρ были депрессия, тревожность, раздражительность, агрессия. Следует отметить, что при метаанализе результатов клинических испытаний на фоне терапии римонабантомρ были отмечены уменьшение окружности талии, снижение систолического и диастолического АД, снижение концентрации триглицерилов и увеличение концентрации ЛПВП. У больных СД 2-го типа наблюдалось достоверное снижение уровня гликемии натощак и HbA1C .
В 2012 г. в США было зарегистрировано два новых препарата центрального действия для лечения ожирения: кьюсимиаρ и бельвикρ , Кьюсимиаρ является комбинированным препаратом, содержащим топирамат и фентермин. Фентермин предсталяет собой симпатомиметический амин, оказывающий аноректический эффект. Под действием фентермина происходит увеличение концентрации норадреналина в гипоталамических центрах. Топирамат, противоэпилептическое средство, увеличивает активность одного из нейромедиаторов - γ-аминомасляной кислоты. Точный механизм действия остается не совсем понятен.
Лоркасеринρ (бельвикρ ) - селективный агонист 5-НТ2с серотониновых рецепторов. Селективная стимуляция 5-НТ2с серотониновых рецепторов, расположенных в ЦНС, повышает чувство насыщения. Терапия неселективными агноистами серотониновых рецепторов, например фенфлюрамином, сопровождается достаточно эффективным снижением массы тела, но сопряжена с развитием побочных эффектов за счет активации 5-НТ2в серотониновых рецепторов, самым серьезным из которых является поражение клапанного аппарата сердца.
В 2014 г. Управление по контролю за пищевыми продуктами и ЛС США одобрило еще один препарат для лечения ожирения - бупропион/налтрексон (контрэйвρ ). Бупропион - ингибитор обратного захвата допамина и норадреналина, обладающий анорексигенным действием, стимулирующий выработку нейронами гипоталамических центров α-меланоцитстимулирующего гормона. Налтрексон - антагонист опиоидных рецепторов, достаточно давно применяется в клинической практике для лечения алкогольной и наркотической зависимости. Налтрексон и его метаболиты, конкурентно связываясь с μ- и κ-опиоидными рецепторами, снижают аппетит и потребление пищи. Монотерапия налтрексоном не сопровождается значимой редукцией массы тела, однако в комбинации с бупропионом анорексигенные эффекты потенциируются. По результатам клинических исследований, дополнительное снижение массы тела на фоне комбинации бупропион/ налтрексон за вычетом эффекта плацебо составляет в среднем 4,5% от исходного значения.
В настоящее время в арсенале врачей в нашей стране существует только два зарегистрированных ЛС: орлистат и сибутрамин, о которых и пойдет речь ниже. Эти препараты имеют разные механизмы действия, применение каждого из них в качестве составной части комплексной программы по лечению ожирения позволяет достичь значимого снижения массы тела. В ближайшей перспективе ожидается регистрация лираглутида, аналога ГПП-1, в качестве ЛС для лечения ожирения. Дозы, эффективные для лечения ожирения, превышают терапевтические дозы, назначаемые при СД 2-го типа. Клинические испытания этого препарата успешно завершены, в ряде стран уже разрешено использование лираглутида в дозе 3 мг/сут в широкой клинической практике.
Орлистат
Орлистат - ЛС периферического действия. При приеме внутрь благодаря сходству орлистата с триглицеридами препарат ковалентно связывает сериновый остаток активного центра молекул желудочной и панкреатической липаз и ингибирует фермент. Связывание носит обратимый характер, но в физиологических условиях подавляющий эффект ЛС в отношении липаз наблюдают на протяжении всего ЖКТ. В результате орлистат нарушает расщепление пищевых жиров и уменьшает их всасывание, а нерасщепленные триглицериды выходят с калом. Препарат снижает всасывание жиров из пищи примерно на 30%. При систематическом применении указанный эффект приводит к уменьшению массы тела у пациентов с ожирением. Системные эффекты у орлистата отсутствуют, поскольку он практически не всасывается в ЖКТ.
ФАРМАКОКИНЕТИКА
Как уже было отмечено, орлистат почти не всасывается в ЖКТ. После однократного приема препарата концентрация жира в каловых массах возрастает через 24-48 ч и приходит к исходному уровню через 48-72 ч. Время полной элиминации орлистата составляет 3-5 сут. Орлистат выводится в основном с калом (97%), в том числе 83% в неизмененном виде, только 2% препарата выделяют почки.
ПОКАЗАНИЯ
Орлистат показан при алиментарно-конституциональном ожирении у пациентов с ИМТ от 30 кг/м2 и более, а также при избыточной массе тела (ИМТ ≥27 кг/м2 ) в сочетании с другими факторами риска развития сердечно-сосудистых заболеваний (СД 2-го типа, дислипопротеинемия и др.).
При приеме препаратов наблюдают значительное снижение уровня общего холестерина, холестерина ЛПНП, повышение уровня холестерина ЛПВП, снижение АД, уровня инсулина натощак, риска развития СД 2-го типа. У пациентов с СД 2-го типа отмечают улучшение показателей углеводного обмена.
Эффективность орлистата оптимальна при приеме ЛС во время или в пределах 1 ч после еды, 30% энергетической ценности которой обеспечивают жиры. Орлистат рекомендуют применять по 1 капсуле (120 мг) 3 раза в день во время еды. Если принимаемая пища не содержит жиров, прием препарата можно пропустить.
Данных о дополнительных преимуществах комбинированной терапии орлистатом и сибутрамином нет.
ПРОТИВОПОКАЗАНИЯ
К противопоказаниям к применению орлистата относят повышенную чувствительность к препарату, синдром хронической мальабсорбции, холестаз, беременность и лактацию.
В настоящее время по риску применения во время беременности орлистат относят к классу В. Адекватных исследований по безопасности орлистата у беременных не проводили. Поскольку данные испытаний на животных нельзя напрямую переносить на человека, орлистат не рекомендуют применять во время беременности. Неизвестно, попадает ли орлистат в грудное молоко, поэтому не следует применять препарат во время лактации.
Безопасность и эффективность использования орлистата у детей не исследованы, поэтому орлистат не рекомендуют назначать детям до 12 лет. Кроме того, ограничениями к применению препарата считают гипероксалурию по данным анамнеза и нефролитиаз (кальциевые оксалатные камни).
ПОБОЧНЫЕ ЭФФЕКТЫ
При приеме орлистата наиболее частыми побочными эффектами бывают частый жидкий и/или жирный стул, маслянистые выделения из прямой кишки, выделение газов с некоторым количеством отделяемого, императивные позывы на дефекацию, недержание кала. Побочные эффекты считают проявлениями действия ЛС. Они дисциплинируют пациента в плане соблюдения рекомендованного режима питания. Ввиду того что орлистат может снижать всасывание жирорастворимых витаминов, во время лечения необходимо ежедневно принимать стандартный комплекс поливитаминов. В отличие от симпатомиметиков, орлистат можно применять длительно. Допустимая продолжительность его применения составляет 4 года.
Препарат не рекомендуют применять во время беременности и кормления грудным молоком. В детском (до 12 лет) и пожилом (после 65 лет) возрасте применение орлистата не изучено.
ВЗАИМОДЕЙСТВИЯ
Никаких клинически значимых отрицательных лекарственных взаимодействий при приеме орлистата не выявлено.
Сибутрамин
В 2010 г. после публикации результатов исследования Sibutramine Cardiovascular OUTcomes, в ходе которого было выявлено повышение частоты нефатальных инсультов и инфарктов у лиц с исходно высоким риском сердечно-сосудистой патологии, препарат меридиа (оригинальный сибутрамин) был отозван компанией-разработчиком с фармацевтического рынка.
В нашей стране Росздравнадзор строго рекомендует назначать препараты сибутрамина только с соблюдением требований Инструкции по медицинскому применению препарата. Противопоказаниями к назначению сибутрамина являются сердечно-сосудистые заболевания [включая ИБС, декомпенсированную хроническую сердечную недостаточность, врожденные пороки сердца, окклюзивные заболевания периферических артерий, тахикардию, аритмию, цереброваскуляр-ные заболевания, а также неконтролируемой АГ (АД выше 145/90 мм рт.ст.)]. Длительность терапии не должна превышать 12 мес. Препарат должен быть отменен при увеличении ЧСС в покое более чем на 10 уд/мин или при увеличении систолического АД более чем на 10 мм рт.ст.
Сибутрамин - анорексигенное средство центрального действия. Препарат ингибирует обратный захват нейромедиаторов, серотонина и норадреналина, из синаптической щели и потенцирует синергические взаимодействия центральных норадреналин- и серотонинергической систем. Сибутрамин снижает аппетит и способствует уменьшению количества потребляемой пищи (усиливает чувство насыщения), а также увеличивает термогенез (вследствие опосредованной активации β-адренорецепторов, отвечающих за липолиз) и оказывает влияние на бурую жировую ткань. ЛС переходит в организме в активные метаболиты (первичные и вторичные амины), значительно превосходящие сибутрамин по способности ингибировать обратный захват серотонина и норадреналина. В исследованиях in vitro активные метаболиты блокируют также обратный захват дофамина, но в три раза слабее, чем норадреналина и 5-гидрокситриптамина. Ни сибутрамин, ни его активные метаболиты не влияют на высвобождение моноаминов и активность моноаминоксидазы, не взаимодействуют с нейротрансмиттерными рецепторами, включая серотонинергические, адренергические, дофаминергические, бензодиазепиновые и глутаматные, не оказывают антихолинергического и антигистаминного действия. Поскольку сибутрамин не имеет выраженного влияния на дофаминергические системы, то не вызывает зависимости и привыкания. Препарат ингибирует захват 5-гидрокситриптамина тромбоцитами и может влиять на функции тромбоцитов.
При снижении массы тела происходит увеличение концентрации в сыворотке крови ЛПВП и снижение количества триглицеридов, общего холестерина, ЛПНП и мочевой кислоты.
Во время лечения наблюдают незначительный подъем АД в покое (в среднем на 1-3 мм рт.ст.) и умеренное увеличение ЧСС (в среднем на 3-7 в минуту), но в единичных случаях возможны более выраженные изменения. Однако при снижении массы тела при длительном применении сибутрамина отмечают снижение АД.
ФАРМАКОКИНЕТИКА
Сибутрамин после приема внутрь быстро всасывается из ЖКТ не менее чем на 77%. При первом прохождении через печень проходит биотрансформацию под влиянием изофермента цитохрома P4503A4 с образованием двух активных метаболитов (моно- и дидесметилсибутрамина). После приема разовой дозы (15 мг) максимальная концентрация монодесметилсибутрамина составляет 4 нг/мл (3,2-4,8 нг/мл), дидесметилсибутрамина - 6,4 нг/мл (5,6-7,2 нг/мл).
Максимальную концентрацию сибутрамина отмечают через 1,2 ч после приема, а через 3-4 ч - максимальную концентрацию активных метаболитов. Одновременный прием пищи снижает максимальную концентрацию метаболитов на 30% и увеличивает время ее достижения на 3 ч. Сибутрамин быстро распределяется по тканям. Связывание с белками составляет 97% (для сибутрамина) и 94% (для моно- и дидесметилсибутрамина). Достижение равновесной концентрации активных метаболитов в крови происходит в течение 4 сут после начала лечения, равновесная концентрация примерно в 2 раза превышает плазменный уровень метаболитов после приема разовой дозы. Период полувыведения сибутрамина составляет 1,1 ч, монодесметилсибутрамина - 14 ч, дидесметилсибутрамина - 16 ч. Активные метаболиты проходят гидроксилирование и конъюгацию с образованием неактивных метаболитов, которые экскретируют преимущественно почки.
ПОКАЗАНИЯ
Сибутрамин показан при алиментарно-конституциональном ожирении у пациентов с ИМТ от 30 кг/м2 и более, а также при избыточной массе тела (ИМТ ≥27 кг/м2 ) в сочетании СД 2-го типа, дислипопротеинемией и др. Ограничения составляет группа лиц, имеющих ИБС, декомпенсированную хроническую сердечную недостаточность, врожденные пороки сердца, окклюзивные заболевания периферических артерий, тахикардию, аритмию, цереброваскулярные заболевания, а также неконтролируемую АГ.
Стартовая доза сибутрамина составляет 10 мг/сут. Если спустя 4 нед потеря массы тела составила менее 2 кг, то при хорошей переносимости дозу можно увеличить до 15 мг/сут. Если спустя еще 1 мес масса тела не снизилась на 2 кг, препарат надо отменить. Прием сибутрамина прекращают, если за первые 3 мес лечения масса тела уменьшилась меньше чем на 5% или после похудения было отмечено увеличение массы тела более чем на 3 кг. При сочетании ожирения с СД 2-го типа, дислипидемией и другими метаболическими заболеваниями лечение сибутрамином можно продолжать и при незначительном снижении массы тела, если таковое связано с улучшением показателей углеводного и/или липидного обмена. Для пациентов с ожирением и СД 2-го типа оптимальная доза составляет 15 мг/сут.
ПРОТИВОПОКАЗАНИЯ
Противопоказаниями к приему сибутрамина считают гиперчувствительность, органические причины ожирения, невротическую анорексию, булимию, психические заболевания, синдром Жиля де ла Туретта, беременность и лактацию, ИБС, декомпенсированную сердечную недостаточность, врожденные пороки сердца, окклюзионные заболевания периферических артерий, нарушения ритма сердца, цереброваскулярные заболевания (инсульт, транзиторные нарушения мозгового кровообращения), АГ (АД более 145/90 мм рт.ст.), гипертиреоз, тяжелые нарушения функций печени или почек, доброкачественную гиперплазию предстательной железы, феохромоцитому, глаукому, установленную лекарственную, наркотическую и алкогольную зависимость, одновременный прием или период менее 2 нед после отмены ингибиторов моноаминоксидазы или других ЛС, действующих на ЦНС (в том числе антидепрессантов, нейролептиков, триптофана), а также других препаратов для уменьшения массы тела.
В настоящее время по риску применения во время беременности сибутрамин относят к классу С, поэтому его использование противопоказано у беременных и кормящих. Ограничениями к применению считают эпилепсию, моторно-вербальный тик (непроизвольные сокращения мышц, нарушение артикуляции), детский и пожилой возраст (безопасность и эффективность применения у детей до 18 лет и у людей старше 65 лет не исследованы).
ПОБОЧНЫЕ ЭФФЕКТЫ
При приеме сибутрамина могут возникать головные боли, бессонница, головокружения, состояния беспокойства, парестезии, изменения вкуса, в единичных случаях судорожные припадки (0,1% случаев), острые психозы. Отмечают тахикардию, сердцебиения, повышение АД, признаки вазодилатации (гиперемию кожи с ощущением тепла). Учитывая потенциальную возможность повышения АД и ЧСС на фоне приема сибутрамина, рекомендуют тщательный мониторинг этих показателей. Перед назначением сибутрамина измеряют АД и проводят ЭКГ. При применении препарата необходим регулярный контроль АД и ЧСС:
Серьезным осложнением при использовании ЛС, повышающих уровень серотонина, считают первичную легочную гипертензию. Это состояние наблюдают крайне редко (у одного из 1 млн пациентов в год), но в половине случаев оно приводит к летальному исходу. Однако установлено, что данное осложнение может быть связано непосредственно с ожирением и курением, а не с приемом сибутрамина.
Со стороны органов ЖКТ в 10% случаев наблюдают сухость во рту, потерю аппетита, запоры, диарею, реже тошноту, обострение геморроя.
Сибутрамин следует с осторожностью назначать при гипокалиемии и гипомагниемии, при нарушениях работы печени и почек легкой и средней степени тяжести. Необходимо иметь в виду, что сибутрамин может уменьшать слюноотделение и способствовать развитию кариеса, заболеваний периодонта, кандидоза полости рта. На период лечения рекомендуют ограничивать употребление алкоголя. Не следует применять препарат во время работы водителями людям, профессия которых связана с повышенной концентрацией внимания.
Женщины детородного возраста в период лечения должны использовать надежные методы контрацепции, поскольку по риску применения во время беременности сибутрамин относят к классу С. Во время беременности и периода лактации применение сибутрамина противопоказано.
ВЗАИМОДЕЙСТВИЯ
При одновременном применении сибутрамина со средствами, обладающими серотонинергической активностью, возрастает риск развития серотонинового синдрома (ажитация, потливость, диарея, повышение температуры тела, аритмии, судороги). Не рекомендуют одновременный прием ЛС, увеличивающих интервал Q-T (астемизол, терфенадин, антиаритмические средства), эфедринсодержащих средств (опасность повышения АД и увеличения ЧСС), а также других анорексигенных препаратов с центральным механизмом действия.
СПИСОК ЛИТЕРАТУРЫ
-
Ожирение: этиология, патогенез, клинические аспекты / Под ред. И.И. Дедова, Г.А. Мельниченко. - М.: Медицинское информационное агентство, 2004. - 456 c.
-
Bray G.A. Drug therapy of obesity. UpToDate, 2007, version 15.1. www.uptodate.com.
-
FDA 2010. Follow-Up to the November 2009 Early Communication about an Ongoing Safety Review of Sibutramine, Marketed as Meridia. 21st Janary 2010.
-
Fleming J.W., McClendon K.S., Riche D.M. New obesity agents: lorcaserin and phentermine/topiramate // Ann. Pharmacother. - 2013. - Vol. 47 (7-8). - P. 1007-1016. doi: 10.1345/aph. 1R779.
-
Gartner R., Haen E. Endokrinpharmakologie. Pharmakotherapie mit Hormonen // Allgemeine und spezielle Pharmakologie und Toxikologie. - 2001. - Vol. 8. - P. 671- 737.
-
Ioannides-Demos L.L., Piccena L. et al. Past, Current and Future Therapies // Journal of Obesity - 2011. Article ID 179674/
-
Li Z., Maglione M., Tu W. et al. Meta-analysis: pharmacologic treatment of obesity // Ann. Intern. Med. - 2005. - Vol. 142. - P. 532.
-
Manning S., Pucci A., Finer N. Pharmacotherapy for obesity: novel agents and paradigms // Ther.Adv. Chronic. Dis. - 2014. - N5 (3). - P. 135-48. doi: 10.1177/2040622314522848.
-
Saper R.B., Eisenberg D.M., Phillips R.S. Common dietary supplements for weight loss // Am.Fam. Physician. - 2004. - Vol. 70. - P. 1731.
-
Scheen A.J. Cardiovascular risk-benefit profile of sibutramine// Am.J. Cardiovasc. Drugs. - 2010. - Vol. 10. - P. 34.
-
Snow V., Barry P., Fitterman N. et al. Pharmacologic and surgical management of obesity in primary care: a clinical practice guideline from the American College of Physicians // Ann. Intern. Med. - 2005. - Vol. 142. - P. 525.