Клиническая фармакология : учебник / В. Г. Кукес, Д. А. Сычев [и др. ] ; под ред. В. Г. Кукеса, Д. А. Сычева. - 6-е изд. , испр. и доп. - Москва : ГЭОТАР-Медиа, 2021. - 1024 с. : ил. - 1024 с. - ISBN 978-5-9704-58815 |
Аннотация
Шестое издание учебника исправлено и дополнено новыми сведениями с учетом последних достижений клинической фармакологии, а также новых государственных образовательных стандартов. Даны современные представления о фармакокинетических процессах, указаны принципы диагностики, коррекции и профилактики нежелательных лекарственных реакций, раскрыты новые механизмы взаимодействия лекарственных средств (на уровне транспортеров). В главе "Основы рациональной фармакотерапии" отражены основные клинико-фармакологические технологии эффективного и безопасного применения лекарственных средств. Клиническая фармакология отдельных групп лекарственных средств рассмотрена с учетом современных представлений о механизме действия, а также с позиций доказательной и персонализированной медицины. Изложена клиническая фармакология новых групп лекарственных средств. Главы, в которых освещены темы, не входящие в примерную программу, размещены в электронной версии учебника, код доступа к которой указан на первом форзаце книги под защитным слоем.
26.3. Антибиотики
β-Лактамные антибиотики
В группу входят препараты, имеющие в структуре р-лактамное кольцо: природные и полусинтетические пенициллины, цефалоспорины, карбапенемы и монобактамы. Вследствие высокой клинической эффективности и низкой токсичности β-лактамные антибиотики составляют основу современной антимикробной терапии, занимая основное место при лечении различных бактериальных инфекций.
Механизм действия р-лактамов заключается в нарушении образования клеточной стенки бактерий за счет необратимого соединения с пенициллинсвязывающими белками микробной стенки делящихся микроорганизмов. Эти белки по своей природе являются ферментами, обеспечивающими синтез бактериальной клеточной стенки. Нарушение их функции вызывает гибель микроорганизмов. В свою очередь, одинаковый механизм действия определяет сходные механизмы резистентности, а также целый ряд общих свойств данных препаратов (бактерицидное действие, синергизм с аминогликозидами, низкую токсичность, возможность перекрестной аллергии у пациентов и т.д.).
Клиническая фармакология пенициллинов
Пенициллин был открыт Александром Флемингом в 1928 г., однако коммерческое производство препарата бензилпенициллина (пенициллина G) стало доступным только в 40-е годы XX в. После идентификации 6-аминопеницилланового ядра было создано большое количество производных, обладающих различными преимуществами, по сравнению с бензилпенициллином (спектр активности, стабильность к действию β-лактамаз, фармакокинетические параметры).
Спектр активности пенициллинов
Спектр антимикробного действия представлен в табл. 26.1 и 26.2.
Природные пенициллины: грамположительные и грамотрицательные кокки (за исключением пенициллиназообразующих штаммов эпидермального и золотистого стафилококка, энтерококка); грамотрицательная микрофлора нечувствительна (за исключением кокков: гонококка, менингококка; палочек: листерий, возбудителя дифтерии, сибирской язвы; спирохет: бледной спирохеты, лептоспиры, боррелии; спорообразующих анаэробов: клостридии; неспорообразующих анаэробов: пептострептококки, фузобактерии); актиномицеты. Пенициллины до сих пор сохраняют высокую активность при стрептококковой и менингококковой инфекции.
В связи с длительным и широким применением спектр действия природных пенициллинов в последнее время сузился за счет селекции штаммов с вторичной резистентностью.
В настоящее время к природным пенициллинам чувствительны только около 10% стафилококков. Пневмококки обладали высокой природной чувствительностью к пенициллину, но в последние 10 лет увеличивается количество штаммов пневмококков, устойчивых к бензилпенициллину.
Пенициллиназоустойчивые пенициллины
Оксациллин - спектр антимикробного действия тот же, что у природных пенициллинов, а также включает пенициллиназообразующие стафилококки; активность оксациллина в отношении пенициллиназообразующего стафилококка выше, чем у пенициллина.
Полусинтетические пенициллины широкого спектра действия (аминопенициллины):
-
ампициллин, амоксициллин - к спектру антимикробного действия природных пенициллинов добавляется активность к энтерококкам, сальмонеллам, шигеллам, протею мирабилис, кишечной палочке и гемофильной палочке;
-
азлоциллинx, пиперациллин (уреидопенициллины) - спектр антимикробного действия тот же, что у ампициллина, а также активность к бактероидам (в высокой концентрации) и части штаммов синегнойной палочки.
Таблица 26 .1. Грамположительная и анаэробная активность пенициллинов

Таблица 26.2. Грамотрицательная активность пенициллинов
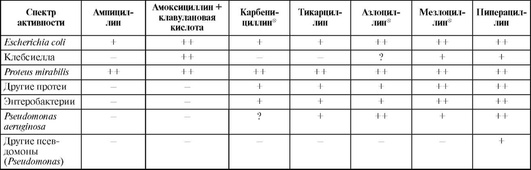
По возрастанию антипсевдомонадной активности «антисинегнойные» пенициллины располагаются в следующей последовательности: карбенициллинx, азлоциллинx, пиперациллин.
Фармакокинетика
Основные фармакокинетические свойства пенициллинов приведены в табл. 26.3-26.5.
Таблица 26.3. Фармакокинетические характеристики пенициллинов
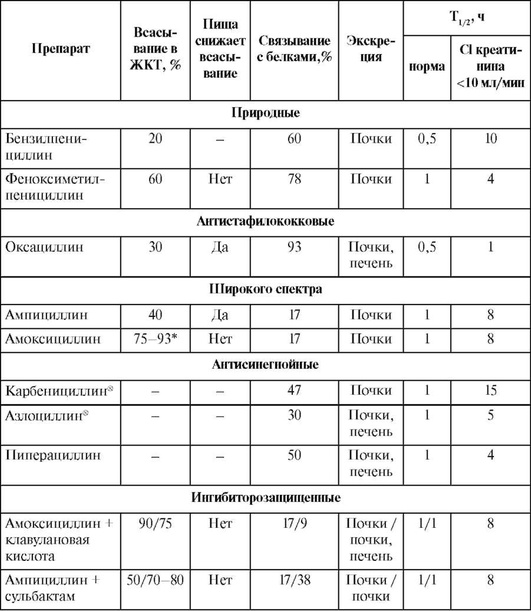
Окончание табл. 26.3
Наиболее высокой биодоступностью (93%) обладают специальные растворимые таблетки амоксициллина - Флемоксин Солютаб.
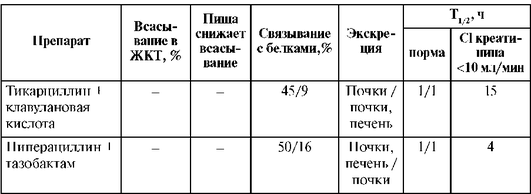
Таблица 26.4. Фармакокинетические параметры полусинтетических пенициллинов
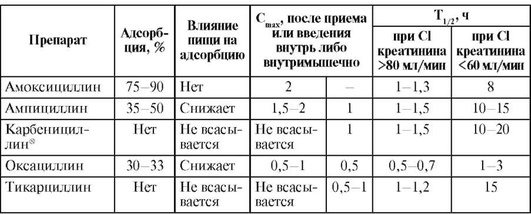
Таблица 26.5. Пути элиминации и метаболизма пенициллинов
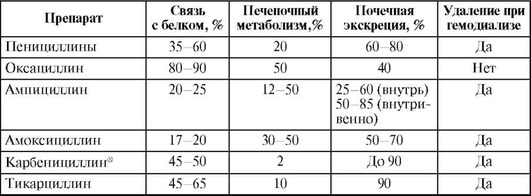
Бензилпенициллин, карбоксипенициллины и уреидопенициллины в значительной степени разрушаются в желудке, поэтому их применяют только парентерально. Феноксиметилпенициллин, оксациллин и аминопенициллины кислотоустойчивы, и их можно назначать внутрь. Наилучшим всасыванием в ЖКТ характеризуется амоксициллин - 75-93%, и его биодоступность не зависит от пищи. Всасывание феноксиметилпенициллина составляет 40-60% (при приеме натощак концентрация в крови несколько выше). Хуже всасываются ампициллин (35-40%) и оксациллин (25-30%), причем пища значительно снижает их биодоступность. Всасывание ингибитора р-лактамаз клавуланата составляет 75%.
Бензилпенициллин прокаина и бензатина бензилпенициллин вводят только внутримышечно. Медленно всасываясь из места инъекции, они создают более низкие, по сравнению с бензилпенициллина Натриевой солью*, концентрации в сыворотке крови. Эти антибиотики обладают пролонгированным действием, поэтому их иногда называют «депо-пенициллинами». Терапевтический уровень бензилпенициллин прокаина в крови сохраняется в течение 18-24 ч, а бензатина бензилпенициллина - 3-4 нед.
Бензилпенициллин применяют внутримышечно, а также его можно вводить внутривенно капельно; при менингитах возможно эндолюмбальное введение натриевой соли.
Cmax бензилпенициллина в сыворотке крови зависит от величины применяемой дозы, однако при введении среднетерапевтической разовой дозы (1 млн ЕД для чувствительных микроорганизмов) его концентрация быстро снижается и уже через 6 ч оказывается ниже средней терапевтической (0,2 ЕД/мл). Для высокочувствительных микроорганизмов средняя терапевтическая концентрация равна 0,06 ЕД/мл (стрептококки группы А, пневмококки). Для бактерицидного действия на малочувствительные микроорганизмы и для преодоления вторичного снижения чувствительности многих штаммов грамположительных микроорганизмов концентрация пенициллина в сыворотке крови должна превышать среднюю терапевтическую концентрацию в 5-10 раз. Это может быть достигнуто введением больших доз препарата (5-50 млн ЕД/сут). Режим введения бензилпенициллина зависит от локализации и тяжести поражения. Суточную дозу препарата, составляющую для взрослых 4 -24 млн ЕД, разделяют не менее чем на 4 инъекции, а при использовании больших доз - не менее чем на 6 инъекций.
Возможно сочетание внутривенного капельного и внутримышечного введения. Бензилпенициллина Натриевую соль* можно вводить в полости для быстрого получения бактерицидного уровня концентрации.
Связь бензилпенициллина с белками невысокая - 35-60%. При парентеральном введении антибиотик быстро и хорошо проникает в легкие, печень, почки, миометрий, несколько хуже - в миокард, костную ткань. Концентрация бензилпенициллина в большинстве тканей существенно ниже, чем в плазме (в 2-3 раза). В серозные и синовиальные полости бензилпенициллин проходит медленно и обнаруживается там в низких концентрациях. В высоких концентрациях препарат содержится в желчи и моче. Через ГЭБ препарат проникает умеренно, у больных менингитом его проницаемость повышается. Высокая проницаемость ГЭБ для бензилпенициллина наблюдается также у недоношенных и новорожденных. Препарат незначительно проникает во внутриглазную жидкость и ткани предстательной железы. Бензилпенициллин хорошо проходит через плацентарный барьер. Его концентрация в крови плода составляет 10-50% от уровня в крови матери. Первые 6 дней жизни ребенка, в связи с незрелостью фильтрационной функции почек, препарат можно вводить 2 раза в сутки.
Природные пенициллины длительного действия (бензилпенициллин прокаина, бензатипенициллин) вводят в виде взвеси только внутримышечно. При применении этих препаратов высокие концентрации препарата в крови не достигаются, поэтому их нельзя использовать при тяжелых острых инфекциях.
Феноксиметилпенициллин - это кислотоустойчивая форма пенициллина, которую можно применять внутрь натощак для лечения нетяжелых инфекционных заболеваний у детей старше 2 лет. Спектр действия такой же, как и у бензилпенициллина, однако концентрация в плазме крови значительно ниже. Концентрация в плазме взрослых после приема внутрь 0,5 г феноксиметилпенициллина аналогична внутримышечному введению 300 000 ЕД бензилпенициллина (1 мг = 1610 ЕД пенициллина). Взрослым назначают по 0,5-1,0 г 4 раза в сутки, возможно его чередование с бензилпенициллином: утром и вечером бензилпенициллин, а днем - 2-3 раза феноксиметилпенициллин.
Оксациллин кислотостабилен, его можно назначать не только парентерально, но и внутрь. Cmax в крови достигается через 1 ч после приема внутрь. Оксациллин имеет большое сродство к белкам плазмы крови (88-95%), он плохо проходит через ГЭБ и слабо проникает в серозные полости.
Ампициллин - это кислотоустойчивый препарат, хорошо всасывающийся при приеме внутрь. Парентеральное применение ампициллина позволяет получить концентрацию в крови в 2-3 раза выше, чем при приеме внутрь. Cmax в крови после приема внутрь отмечается через 1-2 ч. Связь с белком низкая (10-31%).
При внутримышечном и внутривенном введении препарат хорошо проникает в ткани, распределяясь в них равномерно в достаточных концентрациях. При менингите концентрация ампициллина в цереброспинальной жидкости составляет 30-35% от таковой в плазме крови. По остальным фармакокинетическим характеристикам этот антибиотик мало отличается от других полусинтетических пенициллинов.
Амоксициллин по спектру действия практически не отличается от ампициллина; лучше всасывается (биодоступность 95%). В толстой кишке концентрация амоксициллина невелика, и применять его для лечения кишечных инфекций нецелесообразно.
Карбенициллинx - это кислотолабильный препарат, назначаемый парентерально. Хуже амоксициллина проникает в ткани и серозные полости, проходит через ГЭБ. Связь с белком составляет 26-47%. В высоких концентрациях содержится в желчи и моче.
Азлоциллинx и пиперациллин вводят внутривенно. T1/2 их составляет 0,9-1,3 ч и 1 ч соответственно. Метаболизируется около 30% препаратов. Выводятся преимущественно почками в неизмененном виде. Азлоциллин8 хорошо проникает в желчь и бронхиальный секрет, а пиперациллин - еще и в костную ткань. Назначают по 1-2 г 4 раза в сутки.
Распределение
Пенициллины распределяются во многие органы, ткани и биологические жидкости. Они создают высокие концентрации в легких, почках, слизистой оболочке кишечника, репродуктивных органах, костях, плевральной и перитонеальной жидкости. Наиболее высокие уровни в желчи характерны для уреидопенициллинов. В небольших количествах проникают через плаценту и в грудное молоко. Плохо проходят через ГЭБ и гематоофтальмический барьер, а также слабо проникают в предстательную железу. При менингите проницаемость ГЭБ увеличивается.
Метаболизм
Клинически значимой биотрансформации в печени могут подвергаться оксациллин (до 45%) и уреидопенициллины (до 30%). Другие пенициллины практически не метаболизируются и выводятся из организма в неизмененном виде. Среди ингибиторов β-лактамаз наиболее интенсивно метаболизируется клавуланат (около 50%), в меньшей степени - сульбактам (около 25%), еще слабее - тазобактам.
Выведение
Большинство пенициллинов экскретируется почками. Бензилпенициллин выводится почками в неизмененном виде путем фильтрации и секреции. Несмотря на это, препарат можно использовать у больных с легкой и средней степенью почечной недостаточности в стандартных режимах. При клиренсе креатинина ниже 30 мл/мин сокращают количество инъекций (с 4 до 3).
T1/2 составляет в среднем около 1 ч (кроме депопенициллинов); он значительно возрастает при почечной недостаточности. Оксациллин и уреидопенициллины имеют два пути элиминации: почки и билиарную систему. Их T1/2 при нарушении функций почек изменяется в меньшей степени.
Почти все пенициллины полностью удаляются при гемодиализе. Концентрация пиперациллина + тазобактама при проведении гемодиализа снижается на 30-40%.
Нежелательные лекарственные реакции
Обычно пенициллины хорошо переносятся. К наиболее частым НЛР, вызванным пенициллинами, относятся аллергические реакции как немедленного, так и замедленного типа: крапивница, сыпь, отек Квинке, лихорадка, эозинофилия, бронхоспазм, анафилактический шок (чаще возникает при использовании бензилпенициллина). Аллергия является перекрестной ко всем антибиотикам пенициллиновой группы. У некоторых пациентов с аллергией на цефалоспорины может также отмечаться аллергия на пенициллины (15-18%).
При применении аминопенициллинов иногда наблюдается неаллергическая «ампициллиновая» макулопапулезная сыпь (5-10%), которая не сопровождается зудом и может исчезнуть без отмены препарата. «Ампициллиновая» сыпь появляется у 75-100% пациентов с инфекционным мононуклеозом, получающих аминопенициллины, поэтому их нельзя применять при этом заболевании.
Терапевтический диапазон доз природных пенициллинов настолько велик, что позволяет использовать дозы препаратов, составляющие 500 000 ЕД/кг в сутки и более. Бензилпенициллин в этих дозах может оказывать нейротоксическое действие при повышении проницаемости ГЭБ (у новорожденных, при токсикозах, гипоксических состояниях, менингитах).
Может отмечаться неврологическая симптоматика: головная боль, тремор, судороги (чаще у детей и у пациентов с почечной недостаточностью при применении карбенициллина0 или очень высоких доз бензилпенициллина), психические расстройства (при введении высоких доз бензилпенициллина прокаина).
Со стороны ЖКТ могут быть боли в животе, тошнота, рвота, диарея, псевдомембранозный колит (чаще при применении ампициллина и ингибиторзащищенных пенициллинов). При использовании оксациллина в дозах более 6 г/сут или ингибиторзащищенных пенициллинов могут отмечаться НЛР со стороны печени.
При применении карбенициллинаx, высоких доз бензилпенициллина Натриевой соли* может развиться гипернатриемия, сопровождающаяся появлением или усилением отеков у пациентов с сердечной недостаточностью, повышением АД (корригируется применением солей калия).
При парентеральном применении пенициллинов возможно развитие местных реакций: болезненность и инфильтраты при внутримышечном введении, флебиты при внутривенном введении (чаще при применении карбенициллинаx).
Введение депопенициллинов (бензилпенициллин прокаина и бензатина бензилпенициллин) может приводить к развитию сосудистых осложнений: синдрома Онэ (ишемия и гангрена конечностей при введении в артерию), синдрома Николау (эмболия сосудов легких и головного мозга при введении в вену).
Во избежание подобных осложнений следует соблюдать правила применения депопенициллинов:
Возможны гематологические реакции: анемия, нейтропения (чаще при использовании оксациллина); нарушение агрегации тромбоцитов, иногда тромбоцитопения (при применении карбенициллинаx, реже - уреидопенициллинов).
Очень редко со стороны почек может отмечаться транзиторная гематурия (оксациллин) или интерстициальный нефрит.
Пероральные формы (за исключением амоксициллина) лучше принимать натощак.
Показания к применению
Природные пенициллины. В зависимости от особенностей и тяжести течения инфекции возможно применение парентеральных (обычных или пролонгированных) или пероральных лекарственных форм природных пенициллинов при следующих состояниях:
-
при инфекциях, вызванных Streptococcus pyogenes, и их последствиях (тонзиллофарингит, скарлатина, рожа, круглогодичная профилактика ревматизма);
-
инфекциях, вызванных другими стрептококками (инфекционный эндокардит - в комбинации с гентамицином или стрептомицином, профилактика эндокардита в стоматологии - феноксиметилпенициллином), менингококковой инфекции;
Поскольку пролонгированные пенициллины не создают высоких концентраций в крови и практически не проникают через ГЭБ, они не применяются для лечения тяжелых инфекций. Показания к их использованию ограничиваются лечением сифилиса (кроме нейросифилиса), рожистого воспаления, скарлатины, длительной профилактикой ревматической лихорадки.
Феноксиметилпенициллин применяют при лечении легких и среднетяжелых стрептококковых инфекций (тонзиллофарингит, рожистое воспаление).
В связи с нарастанием устойчивости гонококков к пенициллинам его эмпирическое применение при гонорее не оправдано.
Оксациллин применяют при предполагаемой или подтвержденной стафилококковой инфекции различной локализации: инфекции кожи, мягких тканей, костей и суставов, а также при пневмонии.
Аминопенициллины и ингибиторзащищенные аминопенициллины. Основные показания для применения этих препаратов совпадают. Назначение аминопенициллинов более обосновано при легких и неосложненных инфекциях, а их ингибиторзащищенных производных - при более тяжелых или рецидивирующих формах, а также при наличии данных о высокой частоте распространения р-лактамазпродуцирующих бактерий. Путь введения (парентерально или внутрь) выбирают в зависимости от тяжести инфекции. Для приема внутрь более целесообразно использовать амоксициллин или амоксициллин + клавулановая кислота. Препараты данной группы применяются для лечения:
-
бактериальных инфекций верхних и нижних дыхательных путей (средний отит, синусит, обострение хронического бронхита, внебольничная пневмония);
-
внебольничных инфекций мочевыводящих путей (острый цистит, пиелонефрит);
-
менингита, вызванного гемофильной палочкой или Listeria monocytogenes (ампициллин в высокой дозе внутривенно);
-
для лечения и профилактики эндокардита (ампициллин в сочетании с гентамицином или стрептомицином).
Дополнительными показаниями служат инфекции кожи и мягких тканей, интраабдоминальные инфекции, инфекции органов малого таза, периоперационная профилактика в хирургии.
Карбоксипенициллины, уреидопенициллины и их ингибиторозащищенные соединения
Карбоксипенициллины в настоящее время потеряли клиническое значение и практически полностью вытеснены пиперациллином или комбинированными препаратами (тикарциллин + клавулановая кислота, пиперациллин + тазобактам). Уреидопенициллины (азлоциллин8, пиперациллин) в комбинации с аминогликозидами применяются при синегнойной инфекции (в случае чувствительности Pseudomonas aeruginosa). Тикарциллин + клавулановая кислота и пиперациллин + тазобактам используются для лечения тяжелых, преимущественно нозокомиальных, смешанных (аэробно-анаэробных) инфекций различной локализации, в том числе:
Лекарственные взаимодействия
Синергизм в отношении синегнойной палочки проявляют азлоциллин0 и пиперациллин с аминогликозидами и ципрофлоксацином. Оксациллин не следует сочетать с рифампицином ввиду их антагонизма. При сочетании ампициллина с аллопуринолом возрастает риск развития «ампициллиновой» сыпи. Требуется соблюдать осторожность при сочетании антисинегнойных пенициллинов с антикоагулянтами и антиагрегантами ввиду потенциального риска повышенной кровоточивости. Не рекомендуется сочетать с тромболитиками.
Клиническая фармакология цефалоспоринов
Цефалоспорины - один из самых обширных классов антибиотиков. Наиболее распространена классификация цефалоспоринов по поколениям (табл. 26.6). Внутри каждого поколения выделяют препараты для парентерального и перорального применения, препараты с антисинегнойной активностью, препараты с антианаэробной активностью и препараты, имеющие одновременно антисинегнойную и антианаэробную активность.
Таблица 26.6. Классификация цефалоспоринов
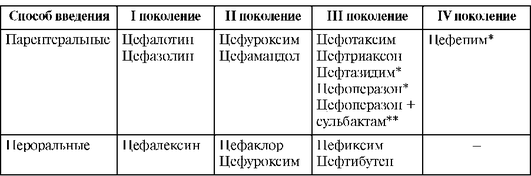
* Препараты с антисинегнойной активностью.
** Комбинация цефоперазона с ингибитором р-лактамаз сульбактамом обладает антисинегнойной и антианаэробной активностью.
Спектр активности
Спектр антимикробной активности цефалоспоринов представлен в табл. 26.7. Следует отметить «пробелы» в спектре активности, характерные для всех цефалоспоринов. Они не активны в отношении метициллинрезистентных стафилококков, энтерококков, листерий, микобактерий, внутриклеточных возбудителей (легионелл, хламидий, микоплазм).
Таблица 26.7. Спектр активности цефалоспоринов
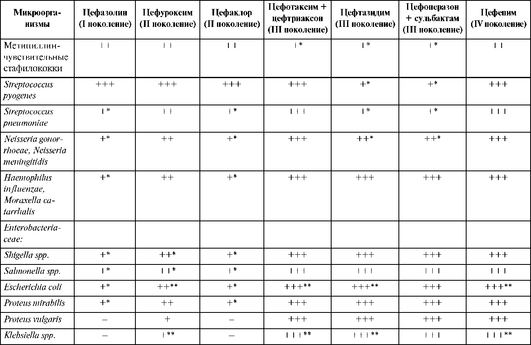
Окончание табл. 26.7
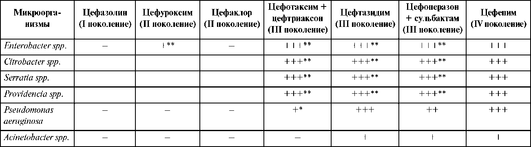
* Обладают активностью in vitro, но не применяются в клинической практике для лечения инфекций, вызванных данным возбудителем.
** Не активны в отношении нозокомиальных штаммов Escherichia coli и Klebsiella pneumoniae, продуцирующих β-лактамазы расширенного спектра действия, Enterobacter spp., Citrobacter spp., Serratia spp. иProvidencia spp., являющихся гиперпродуцентами хромосомных β-лактамаз класса AmpC.
По активности против анаэробов большинство цефалоспоринов обладают умеренной активностью против фузобактерий, пептококков, пептострептококков и не действуют на неспорообразующие анаэробы группы Bacteroides fragilis. Цефалоспорины I поколения имеют узкий спектр антимикробной активности. В современной клинической практике основное значение имеет их активность против метициллинчувствительных стафилококков и стрептококков.
Цефалоспорины II поколения отличаются от цефалоспоринов I поколения более высокой активностью против грамотрицательных микроорганизмов (гонококков, менингококков, гемофил, Moraxella catarrhalis, Escherichia coli, шигелл, сальмонелл, Proteus mirabilis, Proteus vulgaris, клебсиелл), против Streptococcus pneumoniae, а по действию на стафилококки и стрептококки близки к цефалоспоринам I поколения. Следует отметить, что пероральный цефалоспорин II поколения цефаклор менее активен против гемофил и пневмококков, чем цефуроксим. Несмотря на хорошую активность in vitro, цефалоспорины II поколения не применяются для лечения кишечных инфекций и менингита вследствие большей эффективности цефалоспоринов III поколения (цефотаксима и цефтриаксона).
Цефалоспорины III поколения обладают более высокой активностью, чем препараты предыдущих поколений, в отношении грамотрицательных микроорганизмов (гонококков, менингококков, гемофил, Moraxella catarrhalis, представителей семейства Enterobacteriaceae), пневмококков (в том числе пенициллинрезистентных штаммов), высокоактивны против других стрептококков, однако несколько уступают цефалоспоринам I-II поколения по антистафилококковой активности.
Необходимо отметить, что все цефалоспорины III поколения не действуют на представителей семейства Enterobacteriaceae, имеющих механизмы приобретенной резистентности: штаммы Escherichia coli и Klebsiella pneumoniae, продуцирующие β-лактамазы расширенного спектра действия, штаммы энтеробактера, цитробактера, серрации и провиденции, являющиеся гиперпродуцентами хромосомных р-лактамаз класса AmpC.
Среди цефалоспоринов III поколения два препарата (цефоперазон и особенно цефтазидим) обладают клинически значимой антисинегнойной активностью. Однако по активности против стрептококков и пневмококков антисинегнойные цефалоспорины уступают цефуроксиму, цефотаксиму и цефтриаксону.
Цефоперазон + сульбактам (комбинация антисинегнойного цефалоспорина III поколения цефоперазона с ингибитором β-лактамаз сульбактамом) отличается высокой активностью против β-лактамазопродуцирующих микроорганизмов: грамотрицательных бактерий семейства Enterobacteriaceae, ацинетобактера, Bacteroides fragilis и других неспорообразующих анаэробов.
Пероральные цефалоспорины III поколения (цефиксим и цефтибутен) характеризуются более узким спектром активности, чем парентеральные препараты того же поколения. Это прежде всего касается активности против пенициллинрезистентных пневмококков и связано с относительно невысокой биодоступностью и меньшей степенью аффинности к пенициллинсвязывающим белкам у этих препаратов.
Цефалоспорины IV поколения (цефепим) активнее цефалоспоринов III поколения в отношении штаммов Enterobacteriaceae, особенно против энтеробактера, цитробактера, серрации и провиденции, продуцирующих хромосомные AmpC β-лактамазы; Pseudomonas aeruginosa и стафилококков. Цефалоспорины IV поколения по действию на пневмококки, другие стрептококки, анаэробы близки к цефалоспоринам III поколения.
Фармакокинетика
Цефалоспорины для парентерального и перорального применения сильно различаются по своим фармакокинетическим характеристикам(табл. 26.8).
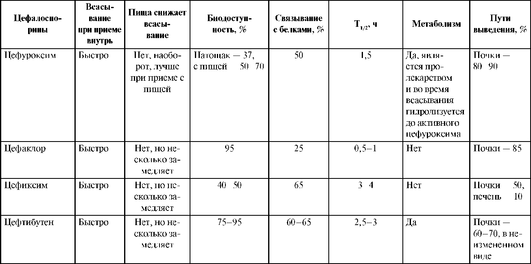
Пероральные цефалоспорины быстро и хорошо всасываются при приеме внутрь, биодоступность составляет от 40-50 (цефиксим) до 95% (цефалексин, цефаклор). Одновременный прием с пищей замедляет скорость всасывания цефаклора, цефиксима и цефтибутена, однако не снижает биодоступность этих препаратов. Цефуроксим (зиннат) является пролекарством, который гидролизуется в ЖКТ с высвобождением активного цефуроксима, причем пища способствует этому процессу.
Цефалоспорины проникают во многие органы, ткани и секреты (в легкие, почки, печень, мышцы, кожу, мягкие ткани, кости, синовиальную, плевральную, перикардиальную и перитонеальную жидкость). Проникают через плаценту. Цефалоспорины III поколения (цефотаксим, цефтриаксон и цефтазидим), а также цефалоспорин IV поколения цефепим хорошо проникают через ГЭБ, и их можно использовать для лечения менингитов. Цефалоспорин II поколения цефуроксим проходит через ГЭБ только при воспалении мозговых оболочек. Хорошо проникают через капсулы абсцессов; на их эффективность не влияют продукты распада тканей. По показаниям, для получения очень высоких концентраций, цефалоспорины можно вводить непосредственно в очаг инфекции.
Таблица 26.8. Фармакокинетические характеристики цефалоспоринов
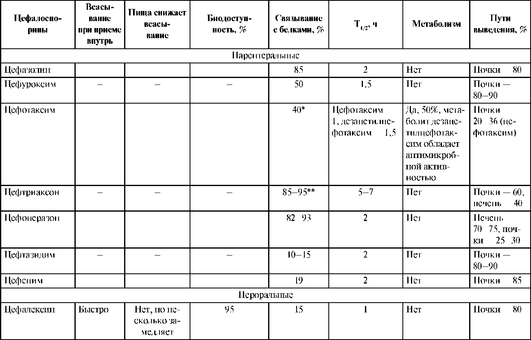
Окончание табл. 26.8
* Связывается с белками плазмы крови на 40%, не вытесняет билирубин из соединения с альбуминами плазмы крови, поэтому предпочтителен у новорожденных.
** Связывается с белками плазмы крови на 85-95%, способен вытеснять билирубин из соединения с альбуминами плазмы крови, поэтому не рекомендуется у новорожденных ввиду риска развития ядерной желтухи.
Большинство цефалоспоринов не метаболизируются. Исключение составляет цефотаксим, 50% введенной дозы которого подвергается биотрансформации с образованием активного метаболита дезацетилцефотаксима, обладающего антимикробной активностью и более длительным (по сравнению с цефотаксимом) T1/2 - 1,5 ч.T1/2 у большинства цефалоспоринов составляет 1-2 ч, что обусловливает необходимость назначения антибиотиков этой группы 3-4 раза в сутки. Более длительный T1/2 (3-4 ч) имеют пероральные цефалоспорины III поколения цефиксим и цефтибутен, и их принимают 1 раз в сутки. Наиболее длительным T1/2 обладает цефалоспорин III поколения цефтриаксон (до 5-7 ч), который при большинстве инфекций применяют 1 раз в сутки, а при менингите - 1-2 раза в сутки.
Большинство цефалоспоринов выделяются почками в неизмененном виде, создавая высокие концентрации в моче. Цефтриаксон и цефоперазон имеют двойной путь выведения (печень и почки).
Нежелательные лекарственные реакции
В целом цефалоспорины хорошо переносятся. Наиболее частая НЛР при применении цефалоспоринов - это аллергия (крапивница, кореподобная сыпь, лекарственная лихорадка, эозинофилия, сывороточная болезнь, анафилактический шок). Возможна перекрестная аллергия между цефалоспоринами и пенициллинами (до 15-18%).
При использовании цефалоспоринов возможны гематологические реакции (лейкопения, гемолитическая анемия и др.), при применении цефоперазона может развиваться гипопротромбинемия со склонностью к кровотечениям и дисульфирамоподобный синдром (повышение чувствительности к алкоголю).
Со стороны ЖКТ возможны боли в животе, тошнота, рвота, диарея, описаны случаи развития псевдомембранозного колита, со стороны печени возможно обратимое повышение активности трансаминаз, возникновение холестаза и псевдохолелитиаза (при применении цефтриаксона).
При назначении парентеральных цефалоспоринов могут возникать местные реакции: болезненность и инфильтраты при внутримышечном введении, флебиты при внутривенном введении.
Показания к назначению
Принимая во внимание различия в спектре антимикробной активности и фармакокинетических показателях цефалоспоринов различных поколений, а также препаратов одного поколения для парентерального и перорального применения, можно выделить следующие основные показания для их назначения.
Цефалоспорины I поколения в настоящее время применяются для лечения инфекций, вызванных стрептококками (но не пневмококками или энтерококками) и метициллинчувствительными стафилококками. Так, цефазолин используют для лечения внебольничных инфекций кожи и мягких тканей, костей и суставов, а также для периоперационной профилактики в хирургии. Основные показания для назначения пероральных цефалоспоринов I поколения - стрептококковый тонзиллофарингит и внебольничные инфекции кожи и мягких тканей, костей и суставов легкой и средней степени тяжести, вызванные чувствительными к ним стрептококками и стафилококками.
Применение цефалоспоринов I поколения при инфекциях мочевыводящих и дыхательных путей в настоящее время нельзя считать рациональным в связи с узким спектром активности, распространением устойчивости среди наиболее вероятных возбудителей и появлением в клинической практике более эффективных антибактериальных препаратов.
Цефалоспорины II поколения можно назначать при всех показаниях, перечисленных для цефалоспоринов I поколения, а также при инфекциях верхних (острый средний отит, острый синусит) и нижних (обострение хронического бронхита, внебольничная пневмония) дыхательных путей, инфекциях мочевыводящих путей (острый цистит, пиелонефрит).
Цефуроксим может служить альтернативой цефазолину при периоперационной антибиотикопрофилактике в хирургии. Цефуроксим успешно используется при проведении ступенчатой терапии.
Цефаклор уступает цефуроксиму по активности в отношении респираторных возбудителей (пневмококков и гемофил), недостаточно хорошо проникает в жидкость среднего уха, поэтому не рекомендуется для лечения острых средних отитов.
Цефалоспорины III поколения назначают для лечения тяжелых внебольничных и нозокомиальных инфекций. Парентеральные препараты без антисинегнойной активности (цефотаксим, цефтриаксон) применяют для лечения тяжелых, угрожающих жизни инфекций, вызванных стрептококками, пневмококками, гемофилами, менингококками, энтеробактериями, таких, как тяжелые формы инфекций дыхательных путей (пневмония, абсцесс легкого и эмпиема плевры - в комбинации с препаратами, обладающими антианаэробной активностью), мочевыводящих путей, инфекций кожи, мягких тканей, костей, суставов, при интраабдоминальных и тазовых инфекциях (в сочетании с антианаэробными препаратами), при генерализованном сальмонеллезе, менингите и сепсисе.
Эти препараты можно использовать для лечения некоторых инфекций в амбулаторной практике, например при острой гонорее (цефтриаксон), а также при остром среднем отите у детей.
Цефалоспорины III поколения с антисинегнойной активностью (цефоперазон, цефтазидим) применяют при инфекциях, вызванных Pseudomonas aeruginosa. Антисинегнойные цефалоспорины III поколения обычно служат одним из обязательных компонентов при комбинированной антибиотикотерапии инфекций на фоне нейтропении. Данные препараты обладают более низкой активностью против Streptococcus pneumoniae, вследствие чего не рекомендуется их использовать при лечении пневмококковых инфекций.
Цефоперазон + сульбактам назначают при тех же показаниях, что и цефоперазон, однако он имеет преимущества при лечении абсцессов легких, эмпиемы плевры, интраабдоминальных и тазовых инфекций вследствие высокой антианаэробной активности, а также инфекций, вызванных бактериями рода ацинетобактер.
Использование цефалоспоринов III и IV поколения для периоперационной профилактики в хирургии нерационально, прежде всего из-за их недостаточной активности против Staphylococcus aureus. Показания к назначению пероральных цефалоспоринов III поколения (цефиксима и цефтибутена) ограничены и включают случаи ступенчатого лечения после применения парентеральных цефалоспоринов III поколения, в случае инфекции мочевыводящих путей, особенно у детей и беременных и кормящих женщин, инфекции дыхательных путей (цефтибутен не рекомендуется при возможной пневмококковой этиологии).
Цефалоспорины IV поколения используют для лечения тяжелых, преимущественно нозокомиальных инфекций, вызванных резистентными возбудителями (энтеробактериями: энтеробактером, цитробактером, серрацией и провиденцией, резистентными к цефалоспоринам II и III поколения за счет гиперпродукции хромосомных AmpC р-лактамаз; а также Pseudomonas aeruginosa), в том числе пневмонии, осложненных инфекций мочевыводящих путей, кожи, мягких тканей, костей, суставов, интраабдоминальных и тазовых инфекций (в сочетании с антианаэробными препаратами), при менингите, сепсисе и нейтропенической лихорадке.
Лекарственные взаимодействия цефалоспоринов
Не рекомендуется смешивать цефалоспорины с другими препаратами в одном шприце или инфузионной системе, необходимо соблюдать рекомендации производителя по использованию определенных растворителей. На всасывание пероральных цефалоспоринов в кишечнике могут влиять пища, одновременный прием антацидов.
При сочетании цефалоспоринов с аминогликозидами или петлевыми диуретиками, особенно у пациентов с нарушениями функций почек, повышается риск развития нефротоксичности.
При сочетании цефоперазона с антикоагулянтами, антиагрегантами и тромболитиками увеличивается риск развития кровотечений. Цефоперазон обладает дисульфирамоподобным эффектом, поэтому недопустимо принимать алкоголь во время лечения этим препаратом.
Клиническая фармакология карбапенемов
В России применяют три антибиотика этой группы: Тиенам* (комбинация имипенема и целастина), меропенем и эртапенем.
Микробиологическая характеристика
Карбапенемы обладают целым рядом микробиологических особенностей, выгодно отличающих их от других р-лактамов:
Спектр активности
Имипенем и меропенем обладают самым широким спектром активности из всех известных в настоящее время антибактериальных препаратов, включая преобладающее большинство клинически значимых грамположительных и грамотрицательных аэробных и анаэробных бактериальных возбудителей (табл. 26.9).
В отношении грамотрицательных микроорганизмов меропенем более активен, чем имипенем, но менее активен в отношении грамположительных микроорганизмов. Карбапенемы высокоактивны в отношении пневмококков, в том числе пенициллинрезистентных штаммов, анаэробов, включая возбудителей интраабдоминальных инфекций.
Таблица 26.9. Спектр активности карбапенемов
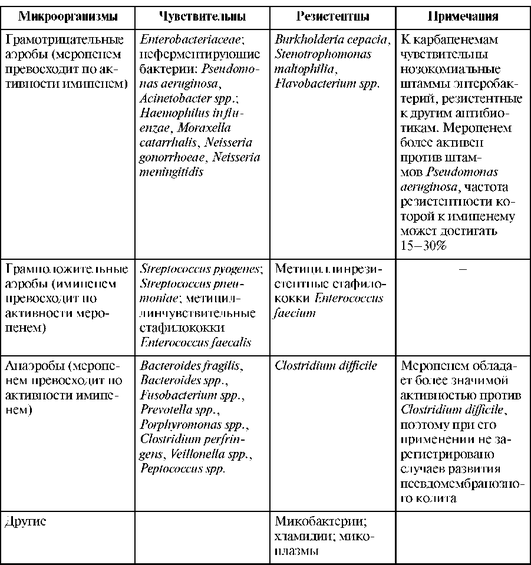
К Тиенаму* устойчивы Xanthomonas maltophilia (ранее Pseudomonas maltophilia), устойчивые к метициллину стафилококки и некоторые штаммы Burkholderia cepacia не чувствительны к имипенему.
Фармакокинетика
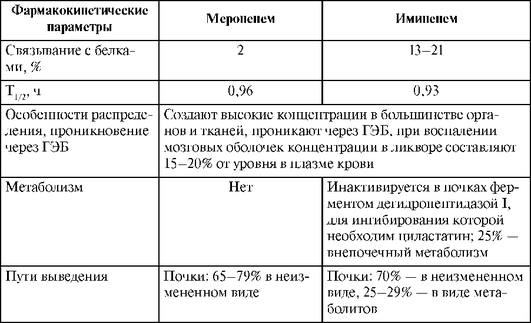
Карбапенемы не всасываются при приеме внутрь. При парентеральном введении имипенем и меропенем по фармакокинетическим параметрам существенно не различаются: T1/2 обоих препаратов составляет почти 1 ч; после введения имипенема в крови создаются несколько более высокие концентрации, по сравнению с меропенемом; в высокой концентрации распределяются в жидкостях и тканях организма, примерно одинаково проникают в спинномозговую жидкость.
Фармакодинамика и фармакокинетика карбапенемов и других β-лактамных антибиотиков схожи.
Карбапенемы выделяются почками путем гломерулярной фильтрации. Одна из особенностей имипенема - гидролиз этого препарата в почках под действием фермента дегидропептидазы I, разрушающей 60-95% введенной дозы. При этом концентрация активного препарата в моче недостаточна для эрадикации возбудителей при инфекциях мочевыводящих путей. Для снижения метаболизма имипенема в почках применяют ингибитор дегидропептидазы I - циластатин, обладающий сходным с имипенемом фармакокинетическим профилем. В настоящее время имипенем применяют в клинической практике в комбинации с циластатином 1:1 под торговым названием Тиенам*. В отличие от имипенема, меропенем устойчив к действию дегидропептидазы I.
Фармакокинетические характеристики карбапенемов представлены в табл. 26.10.
Таблица 26.10. Фармакокинетические характеристики меропенема и имипенема
Нежелательные лекарственные реакции
Карбапенемы характеризуются хорошей переносимостью и низкой частотой развития НЛР. Наиболее часто наблюдаются реакции в месте введения препаратов и реакции со стороны ЖКТ: диарея, тошнота и рвота. Тошнота и рвота чаще возникают при назначении имипенема, поэтому его следует вводить внутривенно в виде длительной капельной инфузии 0,5 г в течение 20-30 мин. При использовании меропенема тошнота и рвота встречаются реже, поэтому его можно вводить внутривенно струйно.
У пациентов с аллергией на β-лактамы возможно развитие перекрестной аллергической реакции немедленного типа к карбапенемам (анафилактический шок, отек Квинке, крапивница и др.).В очень редких случаях при использовании имипенема наблюдают повышение судорожной готовности, что может приводить к возникновению судорог у 0,2-1,5% пациентов, имеющих определенные факторы риска (черепно-мозговая травма, инсульт, эпилепсия, почечная недостаточность, пожилой возраст, превышение рекомендованных доз имипенема). Меропенем не влияет на порог судорожной готовности, что позволяет применять его для лечения менингита. При применении меропенема возможно изменение лабораторных показателей с развитием эозинофилии, нейтропении, лейкопении, анемии, редко - агранулоцитоза, обратимой тромбоцитопении.
Показания к назначению
Широкий спектр антимикробной активности карбапенемов в отношении грамположительных и грамотрицательных, аэробных и анаэробных микроорганизмов делает их препаратами для эмпирической монотерапии тяжелых, угрожающих жизни инфекций:
-
нозокомиальных, тяжелых инфекций полимикробной этиологии (особенно вызванных ассоциациями аэробов и анаэробов) мягких тканей, костей, дыхательных путей, брюшной полости и малого таза;
-
осложненных инфекций мочевыводящих путей, при невозможности применять фторхинолоны;
-
при неэффективной эмпирической терапии тяжелых инфекций у пациентов, получавших другие антибиотики.
Имипенем и меропенем при назначении в одинаковых дозах обладают равной клинической эффективностью. Монотерапия карбапенемами настолько же действенная, а в ряде случаев они превосходят по эффективности традиционно применяемые препараты и их комбинации.
Лекарственные взаимодействия карбапенемов
При приготовлении растворов карбапенемов для парентерального введения следует использовать только рекомендованные производителями растворители. Недопустимо смешивать карбапенемы с другими препаратами в одной инфузионной системе.
Карбапенемы нельзя комбинировать с другими β-лактамами (пенициллинами, цефалоспоринами и азтреонамом) ввиду антагонизма.
При одновременном применении имипенем + циластатина с циклоспорином или теофиллином увеличивается риск развития судорог, особенно у лиц пожилого возраста и пациентов со значительными нарушениями функций почек.
Аминогликозиды
Аминогликозиды относятся к бактерицидным антибиотикам широкого спектра действия. В настоящее время выделяют три поколения аминогликозидов (табл. 26.11).
Таблица 26.11. Классификация аминогликозидов

Клиническая фармакология аминогликозидов
Спектр активности
Стрептомицин, канамицин, виомицинx действуют на Mycobacterium tuberculosis, в то время как амикацин более активен против Mycobacterium avium и других атипичных микобактерий.
Для аминогликозидов II и III поколения характерна дозозависимая бактерицидная активность против грамотрицательных микроорганизмов семейства Enterobacteriaceae (Escherichia coli, Proteus spp., Klebsiella spp., Enterobacter spp., Serratia spp. и др.), а также неферментирующих грамотрицательных палочек (Pseudomonas aeruginosa, Acinetobacter spp.). Аминогликозиды активны против стафилококков, кроме метициллин-резистентных штаммов.
Стрептомицин и гентамицин действуют на энтерококки. Стрептомицин активен против возбудителей чумы, туляремии, бруцеллеза.
Аминогликозиды не активны в отношении Streptococcus pneumoniae, Stenotrophomonas maltophilia, Burkholderia cepacia, анаэробов (Bacteroides spp., Clostridium spp. и др.). Более того, резистентность S. pneumoniae, S. maltophilia и Burkholderia cepacia к аминогликозидам можно использовать для идентификации этих микроорганизмов.
Несмотря на то что аминогликозиды in vitro активны против гемофил, шигелл, сальмонелл, легионелл, клиническая эффективность при лечении инфекций, вызванных этими возбудителями, не была установлена.
Неомицин имеет спектр действия, близкий к стрептомицину, но из-за высокого риска развития НЛР его не вводят парентерально. При приеме внутрь (низкая абсорбция) используется для лечения заболеваний ЖКТ. Применятся наружно при инфекционно-воспалительных заболеваниях кожи.
Фармакокинетика
Фармакокинетические параметры аминогликозидов приведены в табл. 26.12.
Таблица 26.12. Фармакокинетические параметры аминогликозидов
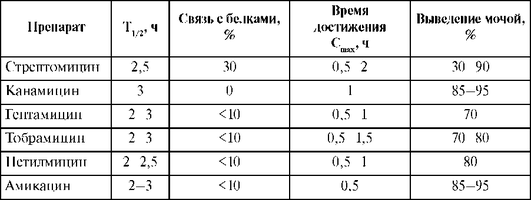
Всасывание
При приеме внутрь аминогликозиды практически не всасываются, внутрь их назначают с целью получения местного эффекта (неомицин). После внутримышечного введения всасываются быстро и полностью. Максимальные (пиковые) концентрации развиваются через 30 мин после окончания внутривенной инфузии и через 0,5-1,5 ч после внутримышечного применения. Основной путь введения парентеральный, можно вводить в серозные полости, эндобронхиально; по жизненным показаниям - эндолюмбально.
Метаболизм
Аминогликозиды не метаболизируются.
Распределение
Пиковые концентрации аминогликозидов варьируют у различных пациентов, поскольку зависят от объема распределения, который, в свою очередь, зависит от массы тела, объема жидкости и жировой ткани, состояния пациента. Например, у пациентов с обширными ожогами, асцитом объем распределения аминогликозидов повышен. Наоборот, при дегидратации или мышечной дистрофии он уменьшается.
Аминогликозиды распределяются во внеклеточной жидкости, включая сыворотку крови, экссудат абсцессов, асцитическую, перикардиальную, плевральную, синовиальную, лимфатическую и перитонеальную жидкость. При сравнении на биологической модели концентрации гентамицина в бронхиальном секрете при внутримышечном (многократном), внутримышечном (однократном) и внутривенном болюсном введении концентрация гентамицина в бронхах достигала уровня минимальной подавляющей концентрации (МПК) только при внутривенном болюсном введении. Аминогликозиды медленно накапливаются в макрофагах (в рибосомах), но при этом антибиотики теряют свою активность. Они способны создавать высокие концентрации в органах с хорошим кровоснабжением: в печени, легких, почках (накапливаются в корковом веществе). Низкие концентрации отмечаются в мокроте, бронхиальном секрете, желчи, грудном молоке. Средняя терапевтическая концентрация обнаруживается в отделяемом ран, гное, грануляциях. В очаги воспаления хорошо проникают только в острой фазе. Аминогликозиды проникают через плаценту, но плохо проходят через ГЭБ. При воспалении мозговых оболочек проницаемость несколько увеличивается. У новорожденных в ликворе достигаются более высокие концентрации, чем у взрослых.
Выведение
Выводятся аминогликозиды в неизмененном виде путем клубочковой фильтрации и тубулярной секреции, создавая высокие концентрации в моче. В течение первых 6-8 ч удаляется 50-80% принятой дозы. Печеночный клиренс имеет значение только для канамицина (его концентрация в желчи составляет 50% от его уровня в крови).
Скорость экскреции зависит от возраста, функционального состояния почек и сопутствующей патологии. У пациентов с лихорадкой она может увеличиваться, а при снижении функционирования почек значительно замедляется. У пожилых экскреция также может замедляться в результате возрастного снижения клубочковой фильтрации. T1/2 всех аминогликозидов у взрослых с нормальным функционированием почек составляет 2-4 ч, у новорожденных - 5-8 ч, у детей - 2,5-4 ч. При почечной недостаточности T1/2 может возрастать до 70 ч и более, и в этом случае необходимо изменение режима дозирования препарата.
Режимы дозирования аминогликозидов
У взрослых пациентов могут осуществляться два режима парентерального назначения аминогликозидов: введение 2-3 раза в сутки (традиционный) и однократное внутривенное капельное введение всей суточной дозы.
При использовании традиционного режима дозирования первоначальная однократная доза гентамицина, тобрамицина и нетилмицина составляет 1-2 мг на 1 кг идеальной массы тела; амикацина и канамицина - 7,5 мг/кг. При нормальной величине клубочковой фильтрации поддерживающую дозу гентамицина, тобрамицина и нетилмицина (3-5 мг/кг в сутки) распределяют на 3 приема. Поддерживающая доза для амикацина и канамицина составляет 15 мг/кг в сутки в 2-3 приема. Продолжительность курса лечения - 7-10 дней; более длительное использование аминогликозидов возможно лишь по очень строгим показаниям.
Однократное внутривенное капельное введение всей суточной дозы аминогликозида позволяет оптимизировать лечение данными ЛС. В ходе многочисленных клинических исследований было подтверждено, что эффективность лечения при однократном режиме назначения аминогликозидов такая же, как и при традиционном, а нефротоксичность выражена в меньшей степени. К тому же при однократном введении суточной дозы снижаются экономические затраты. Однако такой режим назначения аминогликозидов нельзя использовать при лечении бактериального эндокардита.
При выборе дозы аминогликозидов необходимо учитывать такие факторы, как масса тела пациента, локализация и тяжесть инфекции, функции почек.
При парентеральном введении дозы всех аминогликозидов надо рассчитывать на 1 кг долженствующей массы тела. Учитывая, что аминогликозиды плохо распределяются в жировой ткани, у пациентов с массой тела, превышающей идеальную более чем на 25%, необходимо скорректировать дозу. При этом суточную дозу, рассчитанную на фактическую массу тела, следует эмпирически снизить на 25%, а у истощенных пациентов дозу следует на 25% увеличить.
При менингите, сепсисе, пневмонии и других тяжелых инфекциях назначают максимальные дозы аминогликозидов, при инфекциях мочевыводящих путей - минимальные или средние. Максимальные дозы не следует назначать пожилым.
У пациентов с почечной недостаточностью дозы аминогликозидов обязательно необходимо уменьшить либо путем снижения разовой дозы, либо увеличением интервалов между введениями. При сопутствующей почечной недостаточности дозу определяют исходя из концентрации креатинина.
Поскольку фармакокинетика аминогликозидов не стабильна и зависит от целого ряда причин, для достижения максимального клинического эффекта с одновременным снижением риска развития НЛР проводят ТЛМ.
Эффект лечения аминогликозидами зависит от ПК препарата в крови. При фармакокинетическом мониторинге определяют ПК аминогликозида в сыворотке крови через 60 мин после внутримышечного введения или через 15-30 мин после окончания внутривенного.
Пиковая концентрация должна составлять не менее 6-10 мкг/мл для гентамицина, тобрамицина, нетилмицина (до 20 мкг/мл) и не менее 20-30 мкг/мл для канамицина и амикацина (до 50-60 мкг/мл).
Определение остаточной концентрации (перед очередным введением) свидетельствует о степени кумуляции аминогликозида и позволяет контролировать безопасность лечения: менее 2 мкг/мл для гентамицина, тобрамицина, нетилмицина и менее 10 мкг/мл для канамицина и амикацина.
Нежелательные лекарственные реакции
Аминогликозиды обладают потенциальной нефротоксичностью, ототоксичностью и могут вызывать нервно-мышечную блокаду.
Нефротоксический эффект может проявляться значительным увеличением или уменьшением частоты мочеиспускания либо количества мочи, повышенным чувством жажды, снижением клубочковой фильтрации и повышением уровня креатинина в сыворотке крови. К факторам риска нефротоксичности относятся: исходные нарушения функций почек, пожилой возраст, высокие дозы или длительные курсы лечения, одновременное применение других нефротоксичных препаратов (амфотерицина В, полимиксина В, ванкомицина, петлевых диуретиков, циклоспорина).
Ототоксичность проявляется снижением слуха, шумом, звоном или ощущением «заложенности» в ушах. К факторам риска относятся: пожилой возраст, исходные нарушения слуха, высокие дозы или длительные курсы лечения, одновременное назначение других ототоксичных препаратов.
Вестибулотоксичность проявляется нарушением координации движений, головокружением. Она чаще развивается у пациентов пожилого возраста, при исходных вестибулярных расстройствах, использовании высоких доз, длительных курсов лечения.
Нервно-мышечная блокада проявляется угнетением дыхания вплоть до полного паралича дыхательных мышц. Факторы риска: исходные неврологические заболевания (паркинсонизм, миастения), ботулизм, одновременное или предшествующее применение миорелаксантов, нарушение функций почек. Меры помощи: внутривенное введение кальция хлорида или антихолинэстеразных препаратов.
Лекарственные взаимодействия
Аминогликозиды нельзя смешивать в одном шприце или одной инфузионной системе с β-лактамными антибиотиками и гепаринами вследствие физико-химической несовместимости.
В связи с высокой токсичностью аминогликозидов их целесообразно использовать короткими курсами в синергидных сочетаниях с другими антибиотиками. При инфекциях, вызванных синегнойной палочкой, высокоэффективна комбинация аминогликозидов с карбенициллиномx. Синергизм также отмечается при сочетании с бензилпенициллином, цефалоспоринами. Вероятность нефротоксических реакций увеличивается при сочетании с цефалоридиномx, сульфаниламидами, фуросемидом, этакриновой кислотой других препаратов, снижающих тубулярную секрецию. Следует избегать совместного применения с другими нефротоксичными ЛС (полимиксином В, амфотерицином В, ванкомицином). Больные должны получать достаточное количество жидкости для уменьшения повреждающего действия аминогликозидов на почки.
Усиление нервно-мышечной блокады при одновременном применении средств для ингаляционного наркоза, опиоидных анальгетиков, магния сульфата и при переливании большого количества крови с цитратными консервантами.
Индометацин, фенилбутазон и другие НПВС, нарушающие почечный кровоток, могут замедлять скорость экскреции аминогликозидов.
Вероятность аллергических осложнений невысока.
Показания к применению
Основное клиническое значение аминогликозидов - лечение нозокомиальных инфекций, вызванных аэробными грамотрицательными возбудителями, а также бактериального эндокардита. Стрептомицин и канамицин используют при лечении туберкулеза. Неомицин, как наиболее токсичный среди аминогликозидов, применяют только внутрь и местно.
Аминогликозиды не рекомендуется использовать для лечения стафилококковых инфекций, поскольку существуют другие более эффективные, но менее токсичные антистафилококковые препараты.
Аминогликозиды нельзя использовать для лечения внебольничных пневмоний как в амбулаторных, так и в стационарных условиях, что обусловлено отсутствием активности антибиотиков этой группы против основного возбудителя - пневмококка.
Ошибочно назначение аминогликозидов для лечения шигеллезов и сальмонеллезов (как внутрь, так и парентерально), что обусловлено их клинической неэффективностью против возбудителей, локализованных внутриклеточно.
Аминогликозиды не следует применять для лечения неосложненных инфекций мочевыводящих путей, за исключением случаев, когда возбудитель устойчив к другим менее токсичным антибиотикам.
Тетрациклины
К группе тетрациклинов относятся тетрациклин, окситетрациклин, доксициклин, тигециклин.
Тигециклин - это новый антибиотик из группы тетрациклинов. Разработка тигециклина была связана с необходимостью расширения спектра антимикробной активности и преодоления механизмов резистентности бактерий к традиционным тетрациклинам. В первую очередь это касается механизмов резистентности, обусловленных р-лактамазами расширенного спектра, системами активного выведения антибиотиков из клетки (эффлюкса) и мутациями рибосомальных белков.
Общие свойства тетрациклинов:
Клиническая фармакология тетрациклинов
Спектр антимикробной активности
Природный спектр действия тетрациклинов охватывает грамположительные кокки (в настоящее время наблюдается высокая устойчивость пневмококков, стрептококков и большинства стафилококков ко всем препаратам данной группы за исключением тигециклина) и грамотрицательные кокки: гонококки, пневмококки и Moraxella catarrhalis (устойчивы 50% штаммов гемолитического стрептококка и 70% штаммов энтерококков), листерии, возбудители сибирской язвы, иерсинии, хламидии, микоплазмы, кампилобактерии, бруцеллы, гемофильная палочка, Haemophilus influenzae, Haemophilus ducreyi, холерный вибрион, возбудители чумы, туляремии, риккетсии, бледные спирохеты, клостридии (кроме Clostridium difficile), фузобактерии, Propionibacterium acnes (вызывающие развитие угрей). Доксициклин активен в отношении малярийного плазмодия.
Фармакокинетика
Всасывание. При назначении внутрь всасываемость тетрациклина и окситетрациклина составляет 70-80% (табл. 26.13); она снижается при приеме препаратов после еды. Всасывание тетрациклинов происходит медленно. Парентеральное введение создает высокие концентрации препарата в крови, в 2 раза и более превышающие концентрацию после приема внутрь. Наилучшей всасываемостью обладает доксициклин. Биодоступность доксициклина практически не зависит от пищи и составляет 90-95%. Биодоступность тетрациклина натощак - 75%, а при приеме с пищей она снижается в 2 раза. Cmax при приеме внутрь достигается через 1-3 ч; средняя терапевтическая концентрация (1-4 мкг/ мл) поддерживается на протяжении 12-24 ч. Связь с белком составляет 80-95%, T1/2 тетрациклина - 3-6 ч, окситетрациклина - 9-10 ч, а доксициклина - 12-22 ч. Тигециклин вводят внутривенно.
Таблица 26.13. Фармакокинетические параметры тетрациклинов
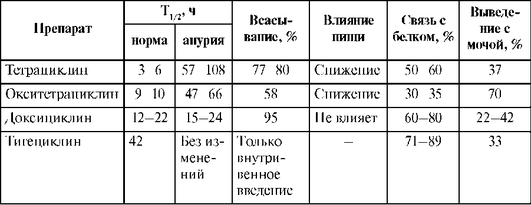
Метаболизм. Тетрациклины метаболизируются в печени. Метаболизм доксициклина значительно выше, чем у тетрациклина и окситетрациклина.
Распределение. Тетрациклины проникают в легкие, печень, почки, селезенку, матку, миндалины, предстательную железу, накапливаются в воспаленной и опухолевой ткани. Доксициклин характеризуется лучшим распределением в ткани. Тетрациклины в комплексе с кальцием откладываются в костной ткани, эмали зубов (особенно в молодой пролиферирующей ткани).
В легочной ткани их концентрация может превышать уровень в крови в 10-15 раз; в желчи (при нормальном функционировании печени) концентрация природных тетрациклинов в 5-10 раз выше, чем в крови. В плевральной жидкости концентрация тетрациклинов составляет 25-75% от наблюдающейся в плазме крови, а в асцитической и синовиальной жидкостях - 50-100% и 60-100% соответственно. Через ГЭБ тетрациклины проникают плохо, даже при парентеральном введении концентрация в цереброспинальной жидкости составляет около 10% от уровня в сыворотке крови, при менингитах - 15-50%. Хорошо проходят через плаценту и накапливаются в грудном молоке. Концентрация тетрациклинов в крови плода и грудном молоке составляет 10-50% и 50-100% от содержания в плазме крови соответственно.
В фармакокинетических исследованиях, проведенных на животных и людях, было показано, что тигециклин быстро распределяется в тканях. В организме человека равновесный объем распределения тигециклина составляет 500-700 л (7-9 л/кг), что подтверждает экстенсивное распределение этого препарата за пределами плазмы и накопление его в тканях. Данные о способности тигециклина проникать в организм человека через ГЭБ отсутствуют.
Выведение. Выводятся тетрациклины в основном с мочой и калом (активная гастроинтестинальная секреция усиливается при заболеваниях почек).
С калом при приеме внутрь в среднем выводится 20-50% принятой дозы, при внутривенном введении - 6-10%. Через почки тетрацикли-ны выводятся путем клубочковой фильтрации. С мочой при приеме внутрь удаляется 10-25% поступившего в организм антибиотика, а при внутривенном введении - 20-70%. При нормальном функционировании печени тетрациклины имеют значительный печеночный клиренс, что приводит к достижению в желчи концентрации, в 5-20 раз превышающей их уровень в крови. Печеночная недостаточность влечет за собой увеличение концентрации тетрациклинов в крови и снижение в желчи. При нарушении выделительной функции почек концентрация тетрациклинов в крови резко возрастает за счет увеличения T1/2.
Доксициклин, в отличие от тетрациклина, характеризуется более высокой липофильностью, поэтому он выводится преимущественно через ЖКТ, причем у пациентов с почечной недостаточностью этот путь основной, и выведение доксициклина не снижается. При гемодиализе тетрациклины выводятся с трудом, а доксициклин не выводится.
59% от назначенной дозы тигециклина выводится через кишечник (при этом большая часть неизмененного тигециклина поступает в желчь), а 33% выводится почками. Дополнительные пути выведения - глюкуронизация и экскреция неизмененного тигециклина почками. Общий клиренс тигециклина после внутривенной инфузии составляет 24 л/ч. На почечный клиренс приходится приблизительно 13% от общего клиренса.
Нежелательные лекарственные реакции
Частота НЛР при использовании тетрациклинов составляет 7-30%. Преобладают токсические осложнения, обусловленные катаболическим действием тетрациклинов (гипотрофия, гиповитаминозы, поражения печени (некрозы), почек (тубулярный некроз), угнетение белкового обмена, ульцерация ЖКТ, фотосенсибилизация кожи (чаще при применении доксициклина), понос, тошнота, повышение внутричерепного давления), биологические осложнения, связанные с подавлением сапрофитов и развитием вторичных инфекций (кандидоз, стафилококковый энтероколит).
Тетрациклины могут вызывать повышение уровня щелочной фосфатазы, амилазы, билирубина, остаточного азота и при лабораторных исследованиях могут дать ложноположительное увеличение уровня катехоламинов в моче.
Тигециклин может повышать АЧТВ, поэтому при его применении рекомендуется проводить мониторинг состояния свертывающей системы крови. Контроль данных показателей необходим также при лечении тигециклином больных, принимающих антикоагулянты (варфарин).
Между тетрациклинами существует перекрестная аллергия.
Внутрь тетрациклины рекомендуется принимать натощак или через 3 ч после еды, запивая 200 мл воды, что уменьшает раздражающее влияние на стенку пищевода и кишечника, улучшает всасывание.
У детей тетрациклины вызывают нарушение образования костной и зубной ткани: дисколорацию зубов (изменение окраски), дефекты эмали, замедление линейного роста костей (поэтому препараты не разрешается применять у детей до 8 лет).
Лекарственные взаимодействия
Пища и антациды значительно снижают биодоступность тетрациклина (но не доксициклина), так как при взаимодействии с содержащимися в пище (особенно в молочных продуктах) и в антацидах катионами кальция, магния и алюминия образуются нерастворимые хелатные соединения.
Карбамазепин, фенитоин и барбитураты ускоряют метаболизм доксициклина в печени и почти в 2 раза укорачивают его T1/2. Аналогичная картина может наблюдаться у лиц, злоупотребляющих алкоголем.
При сопутствующем применении тигециклина и варфарина (в однократной дозе 25 мг) клиренс варфарина снижается. Механизм такого взаимодействия до настоящего времени не установлен.
Показания
-
Инфекции верхних дыхательных путей: острый синусит (доксициклин).
-
Инфекции нижних дыхательных путей: обострение хронического бронхита, внебольничная пневмония (доксициклин, тигециклин).
-
Эрадикация Helicobacter pylori (тетрациклин в сочетании с другими антибиотиками и антисекреторными препаратами).
-
Угревая сыпь при неэффективности местной терапии (доксициклин).
-
Уретрит, вызванный хламидиями, микоплазмами, уреаплазмами (доксициклин).
-
Особо опасные инфекции: чума (в сочетании со стрептомицином), холера (доксициклин).
-
Зоонозные инфекции: лептоспироз, бруцеллез, туляремия (в сочетании со стрептомицином).
Противопоказания
Возраст до 8 лет, беременность, кормление грудью, тяжелая патология печени, почечная недостаточность (тетрациклин).
Макролиды
Макролиды представляют собой класс антибиотиков, основой химической структуры которых служит макроциклическое лактонное кольцо. В клинической практике применяют макролиды, относящиеся к 3 группам (в зависимости от количества атомов углерода в кольце): 14-, 15- и 16-членные (табл. 26.14).
Таблица 26.14. Классификация макролидов

Клиническая фармакология макролидов
Механизм действия
Антимикробное действие макролидов обусловлено нарушением синтеза белка на рибосомах микроорганизма. Макролиды обладают преимущественно бактериостатическим действием, однако в высоких концентрациях они могут действовать бактерицидно на β-гемолитический стрептококк группы А, пневмококк, возбудителей коклюша и дифтерии.
Спектр активности
Макролиды характеризуются высокой активностью против грамположительных кокков, таких как β-гемолитический стрептококк группы А (Streptococcus pyogenes), пневмококк (Streptococcus pneumoniae), золотистый стафилококк (Staphylococcus aureus), за исключением ме-тициллинрезистентных штаммов. Они также действуют на возбудителя коклюша (Bordetella pertussis), дифтерийную палочку (Corynebacterium diphtheriae), моракселлу (Moraxella catarrhalis), легионеллы (Legionella spp.), кампилобактеры (Campilobacter spp.), листерии (Listeria spp.), хламидии (Chlamydia trachomatis, C. pneumoniae), микоплазмы (Mycoplasma pneumoniae), уреаплазмы (Ureaplasma urealyticum). Микроорганизмы семейства Enterobacteriaceae, Pseudomonas spp., Acinetobacter spp. обладают природной устойчивостью ко всем макролидам.
Азитромицин превосходит все другие макролиды по активности в отношении гемофильной палочки (Haemophilus influenzae). Кларитромицин более других макролидов активен против хеликобактера (Helicobacter pylori) и атипичных микобактерий (Mycobacterium avium, M. leprae и др.). Спирамицин активен в отношении некоторых простейших (Toxoplasma gondii, Cryptosporidium spp.). Макролиды обладают высокой активностью в отношении чувствительных штаммов пневмококка in vitro, в то же время концентрация эритромицина и кларитромицина в крови не достигает значений МПК в отношении устойчивых штаммов Streptococcus pneumoniae и Haemophilus influenzae. Так, для азитромицина показатель AUC/МПК по отношению к чувствительным пневмококкам составляет 50, а клиническая эффективность - 94%, в то же время для резистентных пневмококков эти показатели составляют <0,1 и 21% соответственно. У эритромицина и кларитромицина отношение периода превышения МПК антибиотика в плазме крови ко всему периоду между введениями препарата (T >МПК) для Haemophilus influenzae практически равна 0%, а клиническая эффективность не превышает 15-20%. Кроме того, снижение активности макролидов по отношению к Haemophilus influenzae объясняется низкими величинами рН, обычно наблюдаемыми при гнойно-воспалительных процессах в легких и приводящими к дополнительному снижению эффективности макролидов. Несоответствие между концентрацией макролидных антибиотиков и величиной МПК для Streptococcus pneumoniae приводит к росту числа эритромицинрезистентных штаммов. Таким образом, макролиды демонстрируют сравнительно низкую эффективность по отношению к основным возбудителям бактериальной пневмонии, в отличие от высокой эффективности при лечении пневмонии, вызванной атипичными микроорганизмами.
Резистентность микрофлоры к макролидам
Приобретенная резистентность к макролидам может развиваться путем модификации мишени на рибосомах, а также с помощью активного выталкивания (эффлюкс) препаратов из клетки микроорганизма или бактериальной инактивации. При этом полная резистентность микроорганизмов к макролидам, как правило, является перекрестной к 14- и 15-членным макролидам, но не к 16-членным препаратам.
Фармакокинетика
Всасывание. Степень всасывания макролидов в ЖКТ зависит от вида препарата, его лекарственной формы и еды. Пища существенно снижает биодоступность эритромицина, в меньшей степени - других макролидов и практически не влияет на биодоступность кларитромицина и спирамицина. Макролиды относятся к тканевым антибиотикам, поэтому их ПК в сыворотке крови значительно ниже тканевых и варьируют у различных препаратов.
Связь с белками. Связь с белками: эритромицин - 74%, кларитромицин - 70%, рокситромицин - 90%, азитромицин - 23-50%, спирамицин - 10-18%.
Метаболизм. Макролиды метаболизируются в печени при участии цитохрома Р450 с образованием как неактивных метаболитов, так и соединений, обладающих антимикробной активностью.
Распределение. Макролиды хорошо распределяются в организме, создавая высокие концентрации во многих органах и тканях (миндалины, придаточные пазухи носа, легкие, предстательная железа и т.д.). При этом они хорошо проникают внутрь клеток и создают высокие внутриклеточные концентрации. Благодаря высокой способности к диффузии макролиды лучше накапливаются в ткани легкого, достигая там более высоких концентраций, чем в плазме крови. Наиболее показательны в этом плане новейшие макролиды: кларитромицин в дозе 500 мг накапливается в легочной паренхиме в большей концентрации, чем при введении аналогичной дозы эритромицина. Азитромицин обладает примерно такими же свойствами, при этом его концентрация в сыворотке крови обычно определяется с трудом, а в ткани легкого сохраняется на очень высоком уровне в течение 48-96 ч после однократного введения. В общем случае концентрация новых макролидов в слизистой оболочке бронха в 5-30 раз превышает сывороточную. Макролиды лучше проникают в клетки эпителия, чем в жидкость на поверхности эпителия.
Азитромицин после однократного применения внутрь в дозе 500 мг достигает концентрации в выстилающей эпителий жидкости в 17,5 раза большей, чем МПК90 для Streptococcus pneumoniae. Эритромицин при внутривенном введении в дозе 500 мг накапливается в ткани легкого в больших концентрациях, чем при приеме внутрь 1000 мг.
Макролиды плохо проходят через ГЭБ и гематоофтальмический барьер. Они проникают через плаценту и экскретируются в грудное молоко.
Выведение. Метаболиты макролидов выделяются с желчью. Почечная экскреция составляет 5-10%. T1/2 различных препаратов колеблется от 1,5 (эритромицин, джозамицин, мидекамицин) до 65 ч (азитромицин). При почечной недостаточности T1/2 большинства макролидов не изменяется, и коррекции дозы большинства макролидов не требуется, за исключением кларитромицина и рокситромицина, экскреция которых может замедляться. При циррозе печени T1/2 существенно увеличивается.
Нежелательные лекарственные реакции
Макролиды представляют собой одну из самых безопасных групп антибиотиков. Аллергические реакции на них развиваются очень редко. Чаще всего встречаются желудочно-кишечные расстройства - с одинаковой частотой при применении эритромицина, кларитромицина и азитромицина). Это происходит из-за повышения моторной функции вследствие действия макролидов на мотилиновые рецепторы гладкой мускулатуры ЖКТ. Другие препараты переносятся значительно лучше. Умеренная гепатотоксичность препаратов чаще проявляется транзиторным повышением активности печеночных ферментов. Чаще эти явления встречаются при применении эритромицина, в результате длительного использования которого может развиться холестатический гепатит. При внутривенном введении возможно развитие тромбофлебитов из-за местнораздражающего действия.
Лекарственные взаимодействия
Взаимодействия макролидов с другими ЛС обусловлены ингибированием активности микросомальных ферментов печени (система цитохрома Р450). По степени ингибирования макролиды распределяются в следующем порядке: кларитромицин > эритромицин > рокситромицин > азитромицин > спирамицин.
Клинически важное значение имеет взаимодействие эритромицина и кларитромицина с непрямыми антикоагулянтами (варфарин), теофиллином, противосудорожными препаратами (карбамазепин, вальпроевая кислота), циклоспорином, кортикостероидами, дигоксином и другими ЛС, в результате которого повышается риск развития НЛР.
Совместное применение эритромицина и эрготамина может приводить к резким спастическим сосудистым реакциям. Эритромицин увеличивает AUC мидозаламаp (препарат из группы бензодиазепинов), снижает метаболизм блокаторов медленных кальциевых каналов (никардипинx и др.), противоопухолевых препаратов (тамоксифен, винбластин), гиполипидемических ЛС группы ингибиторов ГМГ-КоА-редуктазы (ловастатин, правастатин), снижает метаболизм наркотических анальгетиков (альфентанилp), ингибиторов протеаз (индинавир), что требует тщательного лекарственного мониторинга и коррекции доз.
Сочетание макролидов (кроме спирамицина) с цизапридомx очень опасно ввиду высокого риска развития тяжелых желудочковых аритмий.
Эритромицин, кларитромицин (но не азитромицин или диритромицинx) вызывают увеличение интервала Q-T и повышают риск кардиотоксических проявлении при совместном применении с антигистаминными препаратами (терфенадинx, астемизолx). Антациды замедляют всасывание макролидов из ЖКТ.
Показания
-
Инфекции верхних и нижних отделов дыхательных путей (тонзиллофарингит, синусит, обострение хронического бронхита, внебольничная пневмония).
-
Инфекции, передаваемые половым путем (хламидиоз, уреаплазмоз).
-
Профилактика и лечение микобактериоза, вызванного Mycobacterium avium у больных ВИЧ-инфекцией (кларитромицин, азитромицин).
С профилактической целью макролиды применяют:
Противопоказания
Макролиды противопоказаны лицам с гиперчувствительностью к ним.
Предостережения
При беременности: не рекомендуется применять кларитромицин, рокситромицин, мидекамицин и джозамицин.
Лактация: кормящим женщинам не следует назначать спирамицин и мидекамицин, поскольку они проникают в грудное молоко.
Педиатрия: безопасность применения до 6 мес не определена для кларитромицина.
Нарушение функций почек: при тяжелой почечной недостаточности (клиренс креатинина <30 мл/мин) увеличивается T1/2 кларитромицина и рокситромицина.
Нарушение функций печени: при заболеваниях печени с осторожностью следует применять эритромицин, рокситромицин и джозамицин, при тяжелых заболеваниях печени дозу кларитромицина необходимо снизить.
Растворы эритромицина, кларитромицина и спирамицина обладают выраженным местно-раздражающим действием, поэтому их следует вводить внутривенно капельно. Нельзя вводить макролиды внутривенно струйно и внутримышечно.
Линкозамиды
Группа линкозамидов включает природный антибиотик линкомицин и его полусинтетический аналог клиндамицин, который имеет более высокую активность in vitro. По механизму действия линкозамиды близки к макролидам. Их бактериостатический эффект обусловлен действием на 50S-субъединицу бактериальной рибосомы, что приводит к нарушению синтеза белка. Бактерицидный эффект может проявиться при высоких концентрациях в отношении высокочувствительных микроорганизмов.
Клиническая фармакология линкозамидов
Спектр активности
Линкозамиды активны по отношению к большинству грамположительных кокков. Наиболее чувствительны стафилококки (кроме метициллинрезистентных), стрептококки, пневмококки и неспорообразующие анаэробы - пептококк, пептострептококки, фузобактерии, бактероиды (включая большинство штаммов Bacteroides fragilis). Клиндамицин умеренно активен в отношении некоторых простейших - токсоплазм, пневмоцист, Plasmodium falciparum. Линкозамиды считаются препаратами резерва при стрептококковых и стафилококковых инфекциях, а также при заболеваниях, вызванных неспорообразующими анаэробами.
Существует множество механизмов развития резистентности микроорганизмов к препаратам. Известна быстро развивающаяся перекрестная с макролидами резистентность стафилококков к обоим препаратам.
Фармакокинетика
Всасывание. При приеме внутрь линкомицина всасывается 30-40% препарата; пища замедляет всасывание. Биодоступность линкомицина составляет всего 30%. Клиндамицин лучше всасывается в ЖКТ, его абсорбция и биодоступность достигают 90% и не зависят от пищи. Время достижения Cmax обоих препаратов составляет 2-3 ч. Терапевтическая концентрация сохраняется на протяжении 12 ч. Клиндамицин связывается с белками.
Распределение. Линкозамиды хорошо проникают в различные ткани и органы. Препараты плохо проходят через ГЭБ. В костной ткани линкомицин достигает 15-25%, клиндамицин - до 40% от их концентрации в сыворотке крови. Концентрация в мокроте клиндамицина составляет 30-75%, в желчи, синовиальной и перитонеальной жидкостях - 50%, в плевральной жидкости - до 50-90%, в аппендиксе, фаллопиевых трубах, трофических язвах, ранах и гное - 30%. Линкозамиды хорошо проникают через плаценту и проходят в материнское молоко, составляя 50-100% от уровня в крови.
Метаболизм. Частично метаболизируются в печени (клиндамицин - до 70-80%).
Выведение. T1/2 линкомицина составляет 5-6 ч, клиндамицина - 2-3 ч.
Линкомицин выводится в неизмененном виде и в виде метаболитов в основном с желчью и калом, с мочой экскретируется лишь 10-30% принятой дозы. Концентрация препарата в моче составляет до 17 мкг/мл при приеме внутрь и до 80 мкг/мл - после внутривенного введения. Клиндамицин выводится в течение 4 сут через почки (10%) и кишечник (3,6%) в виде активного препарата, остальное - в виде неактивных метаболитов. Не удаляется с помощью гемодиализа. У больных с заболеваниями печени и почек T1/2 увеличивается.
Нежелательные лекарственные реакции
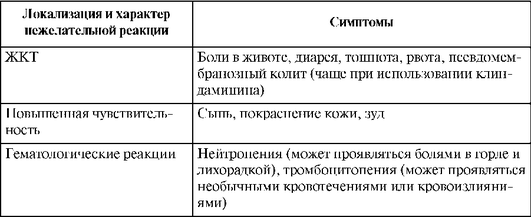
НЛР представлены в табл. 26.15.
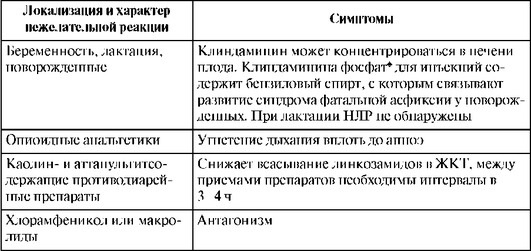
Таблица 26.15. Нежелательные лекарственные реакции линкозамидов
Окончание табл. 26.15
Псевдомембранозный колит может появляться как на фоне приема клиндамицина, так и через 2-3 нед после прекращения лечения. Быстрое внутривенное введение препарата может привести к развитию острой левожелудочковой недостаточности, снижению АД и к остановке сердца.
При назначении клиндамицина в высоких дозах необходим лекарственный мониторинг.
Показания к применению
-
Инфекции нижних отделов дыхательных путей: аспирационная пневмония, абсцесс легкого, эмпиема.
-
Инфекции органов малого таза: эндометрит, аднексит, сальпингоофорит, негонорейный абсцесс маточных труб и яичников, пельвиоцеллюлит, послеоперационные анаэробные вагинальные инфекции.
-
Хлорохинрезистентная тропическая малярия, вызываемая Plasmodium falciparum (только клиндамицин в сочетании с хининомx).
-
Токсоплазмоз (только клиндамицин в сочетании с пириметамином).
При тяжелых инфекциях линкозамиды необходимо сочетать с антибиотиками, действующими на грамотрицательную микрофлору (аминогликозиды и др.).
Противопоказания
Заболевания ЖКТ в анамнезе (неспецифический язвенный колит, энтерит или колит, связанный с применением антибиотиков), повышенная чувствительность к линкозамидам или доксорубицину, беременность, грудное вскармливание. При терминальной почечной или печеночной недостаточности необходимо снизить дозу.
Полипептиды
Полипептиды активны по отношению к грамотрицательной микрофлоре (за исключением кокков): Pseudomonas aeruginosa, Escherichia coli, Haemophilus influenzae, сальмонеллы, шигеллы. Они не действует на большинство штаммов протея, серрации, Bacteroides fragilis, возбудителя туберкулеза, дифтерии, на клостридии и грибы. Влияет лишь на внеклеточно расположенных возбудителей.
Полимиксин В (препарат для парентерального введения) служит средством резерва для лечения синегнойной инфекции: сепсиса, менингита (вводится интралюмбально), пневмонии, инфекций мочевыводящих путей. При инфекциях, вызванных другой грамотрицательной микрофлорой, его используют только при полирезистентности возбудителя к другим менее токсичным препаратам. Не действует на кокковые аэробные и анаэробные микроорганизмы.
Полимиксин Мx (препарат для местного применения) используют для терапии кишечных инфекций и местного лечения синегнойных инфекций раневых поверхностей, отита, язв роговицы.
Клиническая фармакология полипептидов
Фармакокинетика
При применении внутрь практически не всасываются. При инфекционных энтероколитах, дизентерии и для местного лечения раневых поверхностей применяют внутрь.
Полимиксин В при внутримышечном введении всасывается быстро, в крови обнаруживается через 30 мин, максимум его концентрации достигается через 1-2 ч. При повторных введениях в суточной дозе 2-4 мг/кг средняя концентрация составляет 1-8 мкг/мл. Связь с белком незначительная. Препарат плохо проникает через гистогематические барьеры, включая ГЭБ, и быстро инактивируется в гное. Для создания необходимых концентраций необходимо вводить непосредственно в очаги инфекции.
Полимиксин В после внутримышечного введения выделяется с желчью в незначительном количестве. Метаболизируется в печени.
Почками выводится 60% введенной дозы, его концентрация в моче в 20-30 раз больше, чем в плазме крови. При сниженном функционировании почек препарат накапливается в крови.
Нежелательные лекарственные реакции
Нефротоксичность (недопустимо сочетать полипептиды с другими нефротоксичными препаратами), нейротоксичность, проявления нервно-мышечной блокады (особенно у больных с исходной ХПН, миастенией, на фоне применения миорелаксантов), тромбоцитопения, нарушения электролитного баланса (гипокальциемия, гипокалиемия).
Ристомицин
Клиническая фармакология ристомицина
Фармакокинетика
Ристомицинx не всасывается в ЖКТ. Единственный путь его введения - внутривенный (внутримышечно не назначают из-за выраженной болезненности). С белками в плазме крови связывается слабо. Хорошо проникает в лимфу, почки, селезенку, легкие, где его концентрация больше, чем в крови. В меньших концентрациях обнаруживается в печени, миокарде, ткани мозга. При менингитах хорошо проходит через ГЭБ. В плевральных и перитонеальных экссудатах его концентрация в 2-4 раза ниже, чем в крови. Средняя терапевтическая концентрация (5 ЕД/мл) после однократного вливания в дозе 10 000-20 000 ЕД сохраняется более 6 ч.
Кумуляция ристомицинаx может наблюдаться даже при нормальном функционировании почек. Препарат выводится на 80% почками (в основном в течение первых 3 ч после введения). При почечной недостаточности кратность введения уменьшают. С желчью удаляется0,1-0,2%.
Нежелательные лекарственные реакции
При длительном введении развиваются тромбофлебиты, при попадании под кожу образуются болезненные инфильтраты. У 5-12% больных наблюдаются лейкопения, нейтропения (вплоть до агранулоцитоза), анемия, токсическая тромбоцитопения, эозинофилия. Именно поэтому при необходимости использовать препарат в больших дозах, особенно у детей, обязательно 1 раз в 2 дня делать общий анализ крови. Большинство НЛР исчезает через 7-8 дней после отмены препарата. Описано нефро-, гепато- и ототоксическое действие ристомицинаx, а также возможно развитие аллергических реакций.
Гликопептиды
Группа гликопептидов включает два природных антибиотика: ванкомицин и тейкопланин. Рост интереса к гликопептидам обусловлен увеличением частоты нозокомиальных инфекций, вызванных грамположительными микроорганизмами.
Клиническая фармакология гликопептидов
Фармакодинамика
Гликопептиды обладают преимущественно бактериостатическим действием, механизм которого заключается в нарушении синтеза клеточной стенки бактерий.
Спектр активности
Гликопептиды активны в отношении большинства аэробных и анаэробных грамположительных микроорганизмов.
Ванкомицин активен в отношении Staphylococcus aureus (включая пенициллиназообразующие и метициллинрезистентные стафилококки), Streptococcus spp. (включая штаммы, резистентные к пенициллинам), Corynebacterium spp., Haemophilus influenzae. Тейкопланин активен в отношении Staphylococcus aureus, коагулазонегативных штаммов Staphylococcus spp. (включая резистентные к метициллину и другим р-лактамным антибиотикам), Streptococcus spp., Enterococcus faecalis, Listeria monocytogenes, Corynebacterium spp., Clostridium difficile, Peptococcus spp. К нему устойчивы почти все грамотрицательные бактерии, микобактерии, вирусы, простейшие. Перекрестная резистентность с другими антибиотиками отсутствует.
При исследовании in vitro установлены некоторые различия в уровне природной активности и приобретенной резистентности между препаратами: более высокая активность тейкопланина в отношении Staphylococcus aureus (в том числе метициллинрезистентных штаммов), стрептококков (включая Streptococcus pneumoniae) и энтерококков; а ванкомицина - в отношении коагулазонегативных стафилококков.
В последние годы в нескольких странах выделен Staphylococcus aureus со сниженной чувствительностью к ванкомицину или к ванкомицину и тейкопланину.
Некоторые ванкомицинрезистентные штаммы энтерококков сохраняют чувствительность к тейкопланину.
Фармакокинетика ванкомицина
Основной путь введения ванкомицина - внутривенный (внутримышечное назначение очень болезненно). При приеме внутрь он практически не всасывается; этот путь применяют при лечении псевдомембранозного колита.
Средняя терапевтическая концентрация ванкомицина сохраняется в течение 8-10 ч после введения. После внутривенного введения объем распределения составляет 0,4 л/кг, связь с белком - 55%. Препарат практически не метаболизируется. При интраперитонеальном введении в дозе 30 мг/кг концентрация в плазме крови составляет около 10 мг/кг. Около 60% дозы абсорбируется через 6 ч. При многократном введении наблюдается кумуляция. Препарат быстро проникает в полость перикарда, плевры, синовиальную и асцитическую жидкости (концентрация 50-100% от плазменной). При менингитах концентрация ванкомицина в цереброспинальной жидкости составляет 10-20% плазменной. Препарат обнаруживается в желчи (50% от концентрации в крови), с калом выделяется в незначительном количестве. Основной путь выведения - клубочковая фильтрация (90%). Концентрация в моче составляет 100-300 мкг/мл. T1/2 - 4-6 ч у больных с нормальным функционированием почек. 75% дозы экскретируется в первые 24 ч. У больных со сниженным функционированием почек и у пожилых T1/2 достигает 7,5 сут.
Фармакокинетика тейкопланина
Биодоступность тейкопланина после внутримышечного введения 3-6 мг/кг составляет 90%. T1/2 - 40-70 ч, что позволяет назначать его 1 раз в сутки, а после внутривенного введения 3-6 мг T1/2 - 150 ч. Связь с белками плазмы крови - 90-95%. Экскреция почками - 80%.
Гликопептиды не метаболизируются, выводятся почками в неизмененном виде. При почечной недостаточности необходима коррекция доз. Препараты не удаляются с помощью гемодиализа.
Нежелательные лекарственные реакции
НЛР, вызываемые гликопептидами, представлены в табл. 26.16.
Таблица 26.16. Нежелательные лекарственные реакции гликопептидов
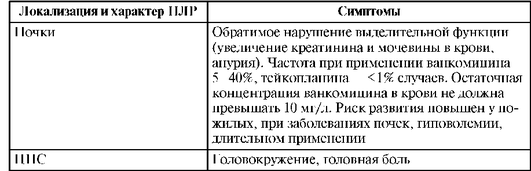
Окончание табл. 26.16
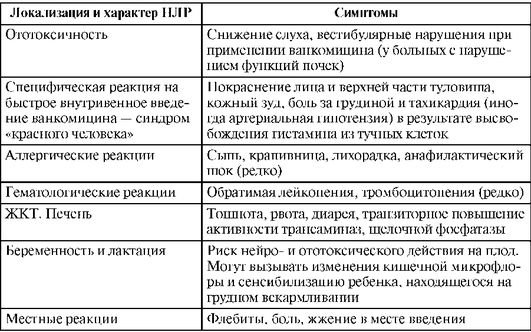
Показания к применению
При инфекциях, вызванных метициллинрезистентными штаммами, а также энтерококками, резистентными к ампициллину и аминогликозидам, гликопептиды служат препаратами выбора.
Показания к применению гликопептидов
-
Тяжелые инфекции, вызванные Enterococcus spp., Corynebacterium jeikeium, Bacillus cereus, Flavobacterium meningosepticum.
-
Инфекционный эндокардит, вызванный зеленящими стрептококками и Streptococcus bovis при аллергии к р-лактамам.
-
Инфекционный эндокардит, вызванный Enterococcus faecalis (в комбинации с гентамицином).
-
Менингит, вызванный Streptococcus pneumoniae со сниженной чувствительностью к пенициллинам.
-
Эмпирическое лечение угрожающих жизни инфекций при подозрении на стафилококковую этиологию:
-
Внутрь - при антибиотикоассоциированной диарее, вызванной Clostridium difficile. Профилактическое применение:
-
периоперационная профилактика при ортопедических и кардиохирургических операциях с высокой частотой распространения метициллинрезистентных штаммов или при аллергии на β-лактамы;
-
профилактика эндокардита у пациентов из групп высокого риска.
Противопоказания
Гиперчувствительность к гликопептидам.
Лекарственные взаимодействия
Лекарственные взаимодействия гликопептидов приведены в табл. 26.17.
Таблица 26.17. Лекарственные взаимодействия гликопептидов
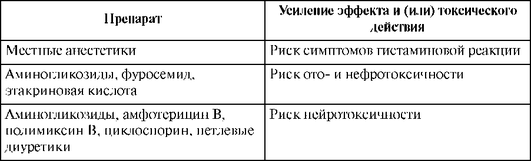
Фузидовая кислота
Так же как гликопептиды, фузидовая кислота - это антибиотик резерва при инфекциях, вызванных штаммами стафилококка, резистентными к пенициллинам и другим антибиотикам.
Фузидовая кислота представляет собой бактериостатический препарат узкого спектра действия. Высокоактивен в отношении Staphyloccocus spp., Neisseria meningitidis, Neisseria gonorrhoeae, менее активен в отношении Streptococcus spp., в том числе S. pneumoniae. Эффективен в отношении стафилококков, устойчивых к пенициллинам, стрептомицинам, хлорамфениколу, эритромицину; Neisseria spp., Haemophilus spp., Moraxella spp., Corynebacteria spp. Препарат обладает слабым противовирусным действием (адено-, риновирусы, вирус осповакцины). Неактивен в отношении грамотрицательных бактерий, а также простейших и грибов.
Клиническая фармакология фузидовой кислоты
Фармакокинетика
Фузидовая кислота эффективна при приеме внутрь, хорошо всасывается (особенно натощак). C в крови достигается через 2-3 ч, средняя терапевтическая концентрация сохраняется на протяжении 24 ч. При повторных приемах может кумулировать. Препарат можно вводить внутривенно. При приеме внутрь в дозе 0,5 г 2 раза в сутки концентрация стабилизируется на уровне 20 мкг/мл, при дозе 1 г 2 раза в сутки - 55-60 мкг/мл. Хорошо проникает в ткани и жидкости организма. Уровень антибиотика в легких, почках, костях, плевральном экссудате, хрящах, соединительной ткани составляет 30-50% от уровня в крови. Благодаря высокой липофильности, в высоких концентрациях обнаруживается в очагах воспаления (40-60% от уровня в крови), в секвестрах. Проникает через плацентарный барьер, в незначительном количестве обнаруживается в молоке матери. Не проходит через ГЭБ. С белками связывается на 90%. Фузидовая кислота выделяется из организма с желчью, где обнаруживается в высоких концентрациях. Метаболизируется в печени до неактивных метаболитов. С мочой выводится лишь 0,1% принимаемого препарата, концентрация в моче составляет 0,3-0,8 мкг/мл. С калом экскретируется 10-15% принимаемого внутрь антибиотика.
Нежелательные лекарственные реакции
Фузидовая кислота - малотоксичный препарат. У 20% больных развивается диспепсия, редко требующая отмены препарата. Иногда наблюдаются аллергические реакции (кожная сыпь, эозинофилия).
Оксазолиденоны
Оксазолиденоны - это новый класс синтетических антимикробных ЛС. В настоящее время в клинической практике применяется один из этих препаратов - линезолид.
Клиническая фармакология оксазолиденонов
Механизм действия оксазолиденонов связан с ингибированием синтеза белка в рибосомах бактериальной клетки. В отличие от других антибиотиков, ингибирующих синтез белка, они действуют на ранних этапах трансляции (необратимое связывание с 30S- и 50S-субъединицей рибосом), вследствие чего нарушается процесс образования 70S-комплекса и формирование пептидной цепи. В результате уникального механизма действия отсутствует перекрестная устойчивость микроорганизмов к линезолиду и другим антибиотикам, действующим на рибосомы (макролиды, линкозамиды, стрептограмины, аминогликозиды, тетрациклины и хлорамфеникол).
Линезолид проявляет высокую активность преимущественно в отношении грамположительных микроорганизмов - стафилококков, энтерококков, пневмококков, различных стрептококков (группы А, В, С и viridans), анаэробных кокков, клостридий и некоторых других микроорганизмов. Большая часть грамотрицательных микроорганизмов природно устойчива к линезолиду. Умеренную активность линезолид проявляет в отношении некоторых грамотрицательных микроорганизмов (МПК90 от 4 мг/л и выше): Moraxella catarrhalis, Haemophilus influenzae, Bordetella pertussis, Neisseria gonorrhoeae, Legionella spp. Критерии чувствительности грамположительных микроорганизмов к линезолиду:
В отношении энтерококков линезолид действует бактериостатически: проявляет стабильную активность в отношении Enterococcus faecalis, Enterococcus faecium и других энтерококков со значением МПК90 от 1 до 4 мг/л, в том числе сохраняет активность в отношении штаммов энтерококков, резистентных к ванкомицину и тейкопланину.
Линезолид проявляет активность (при одинаковых значениях МПК) в отношении метициллинчувствительных и метициллинрезистентных стафилококков, в отношении Staphylococcus aureus и коагулазонегативных стафилококков оказывает бактериостатическое действие.
Линезолид также активен в отношении как чувствительных штаммов Streptococcus pneumoniae, так и штаммов, устойчивых к пенициллинам, эритромицину, цефтриаксону, клиндамицину, тетрациклину, хлорамфениколу. В последние годы выделены штаммы S. pneumoniae со сниженной чувствительностью к линезолиду.
В отношении анаэробных бактерий линезолид проявляет бактерицидный эффект: активность в отношении грамположительных анаэробов (Clostridiumperfringens, Clostridium difficile) и пептострептококков сравнима с активностью ванкомицина в отношении этих микроорганизмов, но в отличие от него линезолид действует также на грамотрицательные анаэробы (Bacteroides fragilis, Fusobacterim spp., Prevotella spp.).
Линезолид высокоактивен в отношении Bacillus spp. (МПК 0,5- 1 мг/л), Corynobacterium spp. (МПК 0,25-0,5 мг/л), Listeria monocytogenes (МПК 0,5-2 мг/л), Mycobacterium tuberculosis (МПК 0,5-2 мг/л), Nocardia spp. (МПК50 2-4 мг/л).In vitro линезолид проявляет слабый постантибиотический эффект. В отношении метициллинрезистентных штаммов, MSSA и ванкомицинрезистентного Enterococcus faecium постантибиотический эффект составляет при одной МПК - 0,5; 0,3; 0,8; при четырех МПК - 0,6; 1,1; 1,4 соответственно. Постантибиотический эффект in vitro увеличивается при повышении дозы ЛС и составляет при дозе 20 и 80 мг/кг в отношении пенициллиночувствительных Streptococcus pneumoniae - 3,6 и 3,8 ч, а в отношении MSSA - 3,9 и 3,7 ч соответственно.
Резистентность к линезолиду развивается медленно. Пока частота выделения штаммов микроорганизмов, устойчивых к линезолиду, существенно меньше, чем к ванкомицину и тейкопланину.
Фармакокинетика
При приеме внутрь линезолид быстро и хорошо всасывается. Биодоступность составляет около 100% и не зависит от приема пищи. Максимальные концентрации в крови достигаются через 1-2 ч, препарат распределяется во многих тканях и средах организма. При многократном применении отношение концентрации в плазме крови к концентрации в бронхиальном секрете и альвеолярной жидкости составляет 1,0:4,5 и 1,0:0,15 соответственно. При однократном применении в случае отсутствия воспаления мозговых оболочек отношение концентрации линезолида в спинномозговой жидкости к концентрации в плазме крови составляет 0,7:1,0. Связь с белком - 31%. Метаболизируется линезолид в печени. Экскретируется преимущественно с мочой, в основном в неактивном состоянии. T1/2 составляет 4,5-5,5 ч, он не зависит от возраста пациента, а также от состояния печени и почек.
Показания
Эффективность линезолида в контролируемых исследованиях установлена при различных инфекциях, вызванных грамположительными микроорганизмами (пневмония, инфекции кожи и мягких тканей, мочевыводящих путей, интраабдоминальные инфекции, эндокардит, сепсис).
Однако он прежде всего показан к применению при инфекциях различной локализации, вызванных мультирезистентными грамположительными бактериями (в первую очередь стафилококками и энтерококками).
Сравнительная клиническая эффективность ванкомицина и линезолида одинакова, но линезолид лучше переносится больными. При инфекциях, вызванных мультирезистентными грамположительными и грамотрицательными бактериями, используются комбинация линезолида с цефалоспоринами III-IV поколения или фторхинолонами.
Хлорамфеникол
Хлорамфеникол - бактериостатический антибиотик широкого спектра активности, нарушает синтез белка в микроорганизме.
Клиническая фармакология хлорамфеникола
Спектр антимикробной активности
Хлорамфеникол эффективен в отношении многих грамположительных и грамотрицательных бактерий, в том числе возбудителей брюшного тифа, дизентерии, менингококковой инфекции, действует на бруцеллы, риккетсии, хламидии, спирохеты, споро- и неспорообразующие анаэробы, грамотрицательные кокки (гонококки, моракселла). На пневмококк, менингококк и гемофильную палочку действует бактерицидно, на другую чувствительную микрофлору - бактериостатически. К хлорамфениколу резистентны микобактерии, клостридии, синегнойная палочка, энтерококки, простейшие, патогенные и простейшие грибы, около 30% штаммов стафилококков. Следует учитывать, что в России 50-90% шигелл и более 10% сальмонелл устойчивы к данному антибиотику. Устойчивость микроорганизмов к хлорамфениколу развивается медленно.
Фармакокинетика
Хлорамфеникол не разрушается в кислой среде желудка, биодоступность составляет 90%, Cmax достигается через 2-3 ч. Терапевтическая концентрация в крови сохраняется 4-5 ч после приема; при применении таблеток с продленным действием - 12 ч. Для быстрого достижения в крови средней терапевтической концентрации (4-10 мкг/мл) применяют внутримышечное и внутривенное капельное введение. Связь с белком - 45-50%.
Основное преимущество хлорамфеникола - высокая проницаемость через гистогематические барьеры. Cmax в ликворе после однократного применения внутрь достигается через 4-5 ч, более низкие концентрации препарата наблюдаются в тканях головного мозга, высокие концентрации содержатся в очагах подострого и хронического воспаления, серозных полостях, в почках, в печени (в желчи обнаруживается до 30% от введенной дозы). В асцитической жидкости концентрация хлорамфеникола может быть выше, чем в крови. Средняя терапевтическая концентрация наблюдается в стекловидном теле, роговице, радужной оболочке, внутриглазной жидкости, в крови плода при приеме его беременной. В грудном молоке уровень препарата в 2 раза меньше. Хлорамфеникол проникает внутрь клетки, что обеспечивает его эффективность при риккетсиозах и бруцеллезе.
Хлорамфеникол подвергается биотрансформации в печени, метаболиты антимикробным действием не обладают. При нарушении функций печени интенсивность его метаболизма замедляется, что способствует удлинению T1/2.
Выведение
T1/2 у взрослых составляет 3,5-5 ч, у детей - 3-6,5 ч.
Выводится хлорамфеникол путем клубочковой фильтрации и ка-нальцевой секреции (10% в активной форме, 90% - в виде водорастворимого неактивного хлорамфеникола глюкуронида). Несмотря на почечный путь экскреции, препарат может применяться у больных с тяжелой почечной недостаточностью, поскольку в этом случае в основном удлиняется время циркуляции в крови его нетоксического метаболита. Опасно, хотя иногда и необходимо, использование хлорамфеникола у новорожденных. Низкая фильтрационная функция почек у новорожденных способствует удлинению времени циркуляции препарата в крови, а недостаточность печеночной глюкуронилтрансферазы обусловливает увеличение концентрации его активной фракции.
Нежелательные лекарственные реакции
Хлорамфеникол - высокотоксичный препарат, и при его применении довольно высока вероятность возникновения токсических осложнений, поскольку они обычно появляются при концентрации препарата в крови выше максимальной терапевтической (25 мкг/мл) дозы. Хлорамфеникол угнетает кроветворение, вызывая гипопластические анемии, лейкопении, тромбоцитопении, ретикулоцитопению. Крайне редко (1 случай на 10-40 тыс. больных) приводит к развитию необратимой, потенциально фатальной апластической анемии, которая может развиться после однократного и даже местного приема через 6-8 нед после отмены препарата.
У новорожденных и недоношенных детей может развиваться интоксикация неметаболизированным хлорамфениколом, так называемый «серый коллапс».
Клинические проявления «серого коллапса» - гипотермия, коллапс, серая окраска кожи, рвота, жидкий стул; летальность составляет40%.
Описаны случаи гепатотоксического и нейротоксического (периферические нейропатии, неврит зрительного нерва) действия хлорамфеникола. При назначении внутрь он также может вызывать диспепсические явления.
Показания
Применение хлорамфеникола ограничено из-за его способности вызывать тяжелые НЛР (в первую очередь гематологические) и вторичной резистентности многих возбудителей. Учитывая высокую частоту и опасность НЛР, хлорамфеникол рассматривают в качестве резервного антибиотика при следующих заболеваниях: бактериальный менингит, абсцесс мозга, интраабдоминальные и тазовые инфекции, генерализованные формы сальмонеллеза, брюшной тиф, риккетсиозы, газовая гангрена.
Взаимодействие
Комбинация хлорамфеникола с пенициллинами и цефалоспоринами целесообразна при сальмонеллезной инфекции; хлорамфеникол также сочетается с макролидами, полиеновыми антибиотиками. При его совместном применении с фенобарбиталом, фенитоином (Дифенин*) и другими индукторами печеночных ферментов ускоряется метаболизм препарата и снижается его концентрация в крови и тканях.
Сульфаниламидные препараты
Сульфаниламидные препараты представляют собой производные сульфаниловой кислоты. Они имеют одинаковый спектр действия, а основные различия между ними определяются особенностью их фармакокинетики.
Клиническая фармакология сульфаниламидных препаратов
Парааминобензойная кислота необходима большинству микроорганизмов для синтеза фолиевой кислоты, которая используется микроорганизмами для образования нуклеиновых кислот. Сульфаниламиды - это структурные аналоги, конкуренты парааминобензойной кислоты. Антимикробная активность сульфаниламидов определяется их сродством к рецепторам микроорганизмов (т.е. способностью конкурировать за рецепторы с парааминобензойной кислотой). При этом опасности повреждения клеток макроорганизма нет, поскольку в них не происходит синтеза фолиевой кислоты (человек получает ее только с пищей). Механизм действия сульфаниламидов объясняет их низкую эффективность в средах с высоким содержанием парааминобензойной кислоты (гной, очаг тканевой деструкции). Большинство микроорганизмов не может утилизировать фолиевую кислоту из окружающей среды, поэтому сульфаниламиды по природной антимикробной активности относятся к препаратам широкого спектра действия. Клиническое применение чаще имеет комбинация с триметопримом. Являясь структурным аналогом птеридиновой части фолиевой кислоты, триметоприм нарушает один из этапов синтеза нуклеиновых кислот - восстановление дигидрофолиевой кислоты в тетрагидрофолиевую (активную форму фолиевой кислоты, ответственную за белковый обмен и деление микроорганизмов) и потенцирует действие сульфаниламидов. Так проявляется синергизм между обоими компонентами ко-тримоксазола, обладающего бактерицидным действием. Применяют следующие комбинированные препараты триметоприма с сульфаниламидами:
Соотношение сульфаниламида и триметоприма 5:1. В отличие от сульфаниламидов, ко-тримоксазол обеспечивает не только бактериостатический, но и бактерицидный эффект.
Спектр действия сульфаниламидов
Сульфаниламиды характеризуются широким спектром антимикробной активности: микроорганизмы (стрептококк, стафилококк, пневмококк, менингококк, гонококк, кишечная палочка, сальмонеллы, холерный вибрион, сибиреязвенная палочка, гемофильная палочка), крупные вирусы (возбудители трахомы, пситтакоза, орнитоза, пахового лимфогранулематоза), простейшие (малярийные плазмодии, токсоплазмы, кокцидии), патогенные грибы (актиномицеты, гистоплазмы).
Умеренно чувствительны энтерококк, стрептококк зеленящий, клебсиеллы, протей, клостридии, пастереллы (в том числе возбудитель туляремии), бруцеллы, микобактерии лепры, лейшмании.
Комбинированные препараты (сульфаниламид + триметоприм) активны в отношении грамположительных и грамотрицательных аэробных кокков: стафилококков (включая некоторые умеренно метициллинрезистентные стафилококки), пневмококков, менингококков, моракселлы, энтеробактерий (Escherichia coli, протеи, шигеллы, сальмонеллы и др.), Haemophilus influenzae (включая некоторые ампициллиноустойчивые штаммы), пневмоцист, токсоплазм и ряда других микроорганизмов.
В настоящее время значительно возросло количество штаммов микроорганизмов, устойчивых к сульфаниламидам, что ограничивает их применение.
К сульфаниламидам устойчивы энтерококки, синегнойная, коклюшная, дифтерийная палочка, микобактерии туберкулеза, бледные спирохеты, лептоспиры и анаэробы.
Фармакокинетика
Всасывание. Основной путь введения сульфаниламидов - пероральный. Существуют парентеральные формы ко-тримоксазола для внутривенного и внутримышечного введения. Сульфаниламиды применяются также наружно в виде мазей или глазных капель. При приеме внутрь всасываются в тонкой кишке, причем скорость и полнота всасывания напрямую зависят от липофильности сульфаниламидов. Ко-тримоксазол хорошо всасывается при приеме внутрь, его биодоступность составляет 90-100%. Cmax в плазме крови достигается через 2-4 ч.
Не всасываются «утяжеленные» сульфаниламиды: сульфагуанидин (Сульгин*), фталилсульфатиазол (Фталазол*), оказывающие при приеме внутрь местное антимикробное действие.
Для уменьшения интенсивности ацетилирования сульфаниламидов в ЖКТ их запивают щелочными растворами.
Связь сульфаниламидов с белками определяет длительность их действия. Связь компонентов (триметоприм и сульфаметоксазол) с белками плазмы крови - 45 и 66% соответственно. Более существенный фактор, влияющий на время циркуляции препарата в крови, - его способность к реабсорбции в почках. Сульфаниламиды, интенсивно связывающиеся с белками плазмы крови, могут конкурировать на этом уровне с другими препаратами.
Все сульфаниламиды хорошо проникают в ткани, причем сульфаниламиды короткого действия проникают быстрее, чем сульфаниламиды пролонгированного действия. Терапевтические концентрации сульфаниламидов обеспечивают бактериостатические концентрации в тканях легких, печени, почек. В плевральной, асцитической и синовиальных жидкостях концентрация сульфаниламидов составляет 50-80% от концентрации в плазме крови. Все сульфаниламиды (кроме сульфадиметоксина) хорошо проникают через ГЭБ, создавая в цереброспинальной жидкости терапевтические концентрации. Концентрация сульфаниламидов длительного действия в желчи продолжительное время выше, чем в плазме.
Метаболизм сульфаниламидов происходит в основном в печени, в меньшей степени - в желудке, кишечнике, почках. Основной путь биотрансформации заключается в ацетилировании. Активность ацетилирования сульфаниламидов зависит не только от свойств препарата, но и от генетических особенностей ферментных систем микроорганизма («быстрые» и «медленные» ацетиляторы). Второй путь биотрансформации сульфаниламидов, реализующийся только в печени, - глюкуронизация. Глюкуроновые метаболиты хорошо растворимы в воде, в почках не реабсорбируются. Их антимикробная активность существенно ниже, чем у неметаболизированных сульфаниламидов. Для сульфадиметоксина глюкуронизация служит основным путем биотрансформации, поэтому при использовании этого препарата отсутствует опасность развития осложнений, связанных с кристаллурией. У новорожденных этот препарат нельзя применять, так как функциональная незрелость глюкуронилтрансферазы приводит к длительной циркуляции и высокой концентрации в крови неметаболизированного препарата, создавая опасность интоксикации.
Выведение. Сульфаниламиды выводятся из организма с мочой в неизмененном виде либо в виде указанных выше парных эфиров и с желчью.
Для лечения инфекции мочевыводящих путей целесообразно назначать сульфаниламиды, выделяющиеся с мочой в активной форме и минимально реабсорбирующиеся, т.е. препараты короткого действия. При ощелачивании мочи увеличиваются ионизация сульфаниламидов, растворимость в воде, снижается реабсорбция, что уменьшает вероятность кристаллурии и способствует поддержанию в моче высоких концентраций сульфаниламидов.
Для обеспечения стойкой щелочной реакции мочи рекомендуется ощелачивающее питье (достаточно назначения соды по 5-10 г/сут), для улучшения растворимости метаболитов сульфаниламидов лечение следует проводить на фоне повышенной водной нагрузки.
Во время лечения нецелесообразно употреблять кислые продукты питания (лимон, клюквенный сок). Эти рекомендации особенно существенны при использовании сульфаниламидов, образующих большое количество плохо растворимых ацетилированных метаболитов, - сульфаниламид (Стрептоцид*), сульфадимидин (Сульфадимезин*), сульфаметоксипиридазин (Сульфапиридазин натрий*), сульфамонометоксинx.
При нарушении выделительной функции почек замедляется экскреция метаболизированных и активных фракций сульфаниламидов, что повышает опасность развития токсических эффектов. При умеренных проявлениях необходимо увеличить интервалы между приемами.
Нежелательные лекарственные реакции
При применении сульфаниламидов могут наблюдаться диспепсические явления (тошнота, рвота) и диарея. Реже отмечаются гематотоксичность (гемолитическая анемия, тромбоцитопения), гепатотоксичность. Особенно часто (у 45-65% пациентов) сыпь и лейкопения отмечаются у пациентов со СПИДом. Высок риск аллергических реакций, синдромов Лайелла и Стивенса-Джонсона. Вероятность токсических и аллергических осложнений сульфаниламидов значительно увеличивается при снижении фильтрационной функции почек.
Одно из наиболее опасных осложнений при применении сульфаниламидов - кристаллизация ацетилированных метаболитов в почках и мочевыводящих путях. У новорожденных и грудных детей сульфаниламиды могут вызывать метгемоглобинемию за счет окисления фетального гемоглобина. Опасно использование сульфаниламидов при гипербилирубинемии, поскольку они, вытесняя билирубин из связи с белками, могут способствовать реализации его токсического действия.
При применении ко-тримоксазола могут развиваться признаки недостаточности фолиевой кислоты (нарушение кроветворения, гипотрофия, поражения ЖКТ). Для лечения этих осложнений может применяться фолиевая кислота.
Показания
Сульфаниламиды могут применяться при кишечных инфекциях: шигеллезе, сальмонеллезе, диарее путешественников, однако большинство шигелл и сальмонелл резистентны к ко-тримоксазолу.
Противопоказания
Сульфаниламиды не следует назначать при аллергии на сульфаниламидные препараты, фуросемид, тиазидные диуретики, ингибиторы карбоангидразы, препараты сульфонилмочевины, при беременности (особенно в I и III триместрах), у детей до 2-месячного возраста. Конкуренция сульфаметоксазола с билирубином за связывание с белками плазмы крови и высокие концентрации свободного сульфаметоксазола повышают риск развития ядерной желтухи у новорожденных. Проникая в грудное молоко, сульфаметоксазол может вызвать ядерную желтуху у детей, находящихся на грудном вскармливании, а также гемолитическую анемию у детей с дефицитом Г-6-ФД. Триметоприм нарушает метаболизм фолиевой кислоты. Допустимо применение ко-тримоксазола у детей с 4-6-недельного возраста, родившихся у ВИЧ-инфицированных матерей.
Сульфаниламиды противопоказаны при тяжелой почечной недостаточности (ко-тримоксазол не следует применять при клиренсе креатинина <20 мл/мин), при тяжелых нарушениях функций печени. Также они не применяются при мегалобластической анемии, связанной с дефицитом фолиевой кислоты.
Лекарственные взаимодействия
Лекарственные взаимодействия представлены в табл. 26.18.
Таблица 26.18. Лекарственные взаимодействия ко-тримоксазола

При хронических воспалительных заболеваниях кишечника (неспецифический язвенный колит, болезнь Крона и др.) эффективна комбинация сульфаниламидов длительного действия с производными аминосалициловой кислоты - месалазином, салазодиметоксином8.
Эти препараты, практически не всасываясь, расщепляются в просвете толстой кишки до сульфаниламидов (сульфапиридазин, сульфадиметоксин), обеспечивающих антимикробное действие, и остатка аминосалициловой кислоты, оказывающей противовоспалительный эффект.
Эффективность сульфаниламидов снижается под действием препаратов, в процессе метаболизма которых образуется парааминобензойная кислота (бензокаин, прокаин, прокаинамид). Антагонистом ко-тримоксазола является фолиевая кислота. Также нецелесообразно на фоне применения сульфаниламидов употреблять пищевые продукты, содержащие в большом количестве парааминобензойную кислоту, в том числе зеленые части растений (цветная капуста, шпинат, морковь, помидоры, бобовые).
Хинолоны и фторхинолоны
Хинолоны - это синтетические антибактериальные препараты. Существует четыре поколения хинолонов, причем три последних являются фторированными (фторхинолоны). Классификация препаратов и спектр антимикробной активности представлены в табл. 26.19.
Таблица 26.19. Классификация и антимикробный спектр хинолонов и фторхинолонов
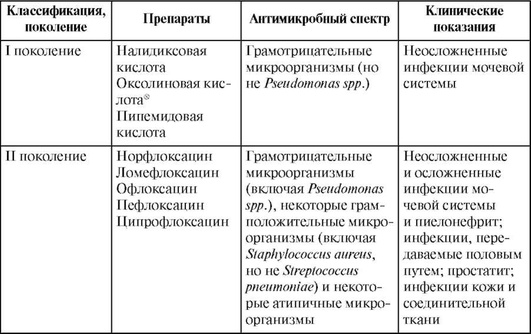
Окончание табл. 26.19
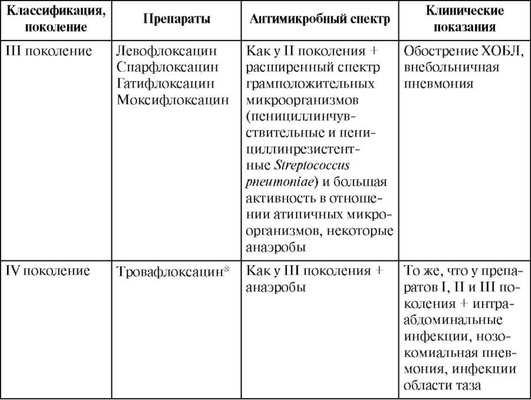
Клиническая фармакология хинолонов и фторхинолонов
Хинолоны
Хинолоны I поколения характеризуются узким спектром активности, и многие из них (оксолиновая кислотаx и пипемидовая кислота) в настоящее время утратили свое значение.
Налидиксовая кислота - это первый 4-хинолон. Этот препарат был синтезирован в 1962 г. Действует бактерицидно, в низких концентрациях возможно также бактериостатическое действие. К налидиксовой кислоте высокочувствительны грамотрицательные бактерии (кроме синегнойной палочки), а грамположительные - обладают природной устойчивостью.
Фармакокинетика хинолонов
При приеме внутрь препарат хорошо всасывается. В печени после гидроксилирования образуется активная форма - гидроксиналидиксовая кислота, связывающаяся в крови на 80-90% с белками, что обусловливает плохое проникновение препарата в ткани. Антимикробным действием обладает только свободная фракция, накапливающаяся в достаточной концентрации лишь в мочевыводящих путях. Активность усиливается при ощелачивании мочи. При сохраненной функции почек терапевтическая концентрация в моче определяется на протяжении 4-6 ч, что объясняет назначение суточной дозы (60 мг/кг) в 4 приема. За сутки выводится 90% препарата, при отсутствии почечной недостаточности препарат не кумулирует.
Фторхинолоны
Фторхинолоны обладают бактерицидным действием, инактивируя ферменты ДНК-гиразу и топоизомеразу IV, нарушая тем самым синтез ДНК в микроорганизме; ломефлоксацин блокирует топоизомеразу II и IV.
Спектр активности фторхинолонов
Фторхинолоны характеризуются значительно более широким антимикробным спектром, высокой бактерицидной активностью, хорошим распределением в органы, что позволяет применять их для лечения широкого спектра инфекций различной локализации (см. табл. 26.19).
Для фторхинолонов характерно действие на грамотрицательные бактерии: кишечную палочку (Escherichia coli), шигеллы, сальмонеллы, протей (Proteus spp.), клебсиеллу (Klebsiella spp.). К ним чувствительны стафилококки (в том числе пенициллинрезистентные и некоторые метициллинрезистентные штаммы), грамотрицательные кокки (гонококк, менингококк, Moraxella catarrhalis), грамотрицательные бактерии семейства Enterobacteriaceae (Escherichia coli, сальмонеллы, шигеллы, протей, энтеробактер, клебсиелла, серрация, провиденция, цитробактер, морганелла), синегнойная палочка (Pseudomonas aeruginosa), кампилобактеры, легионеллы, Mycobacterium tuberculosis. К хинолонам II поколения малочувствительны большинство стрептококков (в том числе пневмококк), энтерококки, хламидии, микоплазмы. Они не действуют на спирохеты, листерии и большинство анаэробов.
Хинолоны III поколения обладают более высокой активностью в отношении как пневмококков, включая пенициллинрезистентные, так и атипичных возбудителей (хламидии, микоплазмы).
Хинолоны IV поколения по антипневмококковой активности и действию на атипичных возбудителей превосходят хинолоны предшествующих поколений. Обладают высокой активностью против неспорообразующих анаэробов (Bacteroides fragilis и др.).
Фторхинолоны III поколения (респираторные фторхинолоны) рекомендованы при лечении внебольничных пневмоний, что объясняется несколькими причинами:
-
респираторные фторхинолоны представляют собой препараты широкого спектра действия, потенциально активные по отношению ко всем наиболее часто встречающимся возбудителям внебольничных пневмоний (см. табл. 26.19): грамположительным микроорганизмам (пневмококки, стафилококки), большинству грамотрицательных и внутриклеточных возбудителей, а также микобактериям туберкулеза и некоторым анаэробам;
-
резистентность Streptococcus pneumonia и других частых возбудителей внебольничных пневмоний к препаратам этой группы за последнее десятилетие практически не изменилась (хотя и наблюдается такая тенденция). В то же время доля пенициллинрезистентных штаммов пневмококка в ряде стран возросла в 5-10 раз. Кроме того, пока нет оснований прогнозировать быстрое увеличение количества микроорганимов, невосприимчивых к действию фторхинолонов, так как возникновение устойчивости определяется мутацией сразу двух различных участков ДНК микроорганизма, а вероятность такого события достаточно низка;
-
многие респираторные фторхинолоны доступны как в формах для внутривенного введения, так и для приема внутрь, что позволяет проводить ступенчатую терапию;
-
большинство респираторных фторхинолонов обладают выгодными фармакокинетическими особенностями, позволяющими применять эти антибактериальные препараты 1-2 раза в сутки.
Наряду с этим приходится констатировать, что стоимость респираторных фторхинолонов существенно выше стоимости антибактериальных препаратов, применяемых в рутинной практике, и кроме того, сохраняется запрет на использование препаратов этой группы для лечения детей и беременных.
Фармакокинетика хинолонов и фторхинолонов
Фармакокинетические параметры представлены в табл. 26.20, 26.21. Фторхинолоны привлекательнее хинолонов и с фармакокинетической точки зрения:
Таблица 26.20. Фармакокинетические параметры хинолонов при приеме внутрь
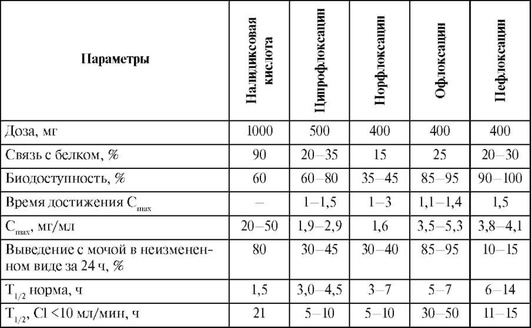
Таблица 26.21. Фармакокинетика фторхинолонов
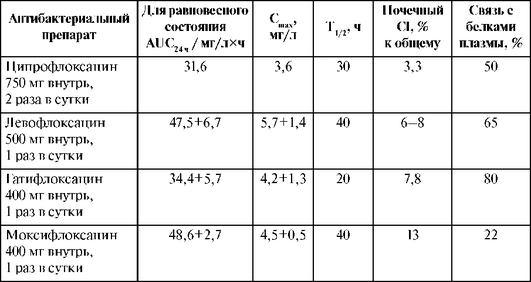
Метаболизм хинолонов и фторхинолонов
В печени препараты данной группы подвергаются метаболизму в различной степени: наиболее активно метаболизируется налидиксовая кислота, оксолиновая кислотаx, пефлоксацин, наименее - пипемидовая кислота, офлоксацин и ломефлоксацин. Препараты группы фторхинолонов ингибируют CYP1A2, осуществляющий деалкилирование теофиллина, и они уже через сутки совместного применения могут повышать концентрацию аминофиллина (Эуфиллин*).
По силе влияния на фармакокинетику теофиллина фторхинолоны подразделяют на три группы.
-
I группа - препараты с выраженным влиянием на фармакокинетику теофиллина и с высоким риском развития НЛР: эноксацинx (через сутки может повысить концентрацию теофиллина на 50-65% с развитием симптомов передозировки теофиллина и даже интоксикации; из-за высокого риска НЛР препарат снят с применения).
-
II группа - препараты, вызывающие умеренное повышение содержания теофиллина в плазме крови (до 40%): ципрофлоксацин (25-30%), пефлоксацин, норфлоксацин (10-15%).
-
III группа - препараты, не взаимодействующие с теофиллином на уровне метаболизма: офлоксацин, ломефлоксацин, спарфлоксацин.
Распределение хинолонов и фторхинолонов
Хинолоны I поколения не создают терапевтических концентраций в крови, во многих органах и тканях, образуя при этом терапевтически значимые концентрации в моче.
Фторхинолоны характеризуются большим объемом распределения, создают высокие концентрации во многих органах и тканях. Все эти препараты примерно одинаково хорошо поступают в ткань легкого. Более высокие концентрации фторхинолонов (для одних и тех же доз) достигаются при внутривенном введении. Фторхинолоны накапливаются в слизистой оболочке бронхов примерно в той же концентрации, что и в плазме крови, что нельзя сказать ни об одном из β-лактамных препаратов (их концентрация в ткани легкого всегда меньше, чем в крови). Концентрация фторхинолонов в эпителиальной жидкости очень высокая. Фторхинолоны способны проникать внутрь клеток, но более кислая внутриклеточная среда приводит к усилению ионизации, поэтому наблюдается обратный поток препарата из клеток наружу. Однако в целом у препаратов этой группы внутриклеточные концентрации превышают внеклеточные, что согласуется с доказанной в ходе клинических исследований эффективностью пефлоксацина и ципрофлоксацина при пневмонии, вызываемой Legionella pneumophila. Ципрофлоксацин, офлоксацин и пефлоксацин проникают через ГЭБ.
Выведение хинолонов и фторхинолонов
Выводятся из организма преимущественно почками, частично с желчью. При нарушении функций почек выведение препаратов замедляется, особенно офлоксацина и ломефлоксацина. При тяжелой почечной недостаточности необходима коррекция доз всех фторхинолонов. При проведении гемодиализа фторхинолоны удаляются из крови в незначительном количестве (10-30%).
Нежелательные лекарственные реакции
При применении хинолонов наиболее часто наблюдается гепатотоксическое действие. Могут возникать диспепсии (особенно при приеме натощак), изредка развиваются аллергические реакции, симптомы поражения ЦНС (головокружение, головная боль, повышение судорожной активности), панцитопения и гемолитическая анемия.
Препарат противопоказан новорожденным и детям первых месяцев жизни, а также больным с заболеваниями печени.
Для фторхинолонов характерно торможение развития хрящевой ткани, поэтому они противопоказаны беременным и кормящим матерям; у детей могут применяться только по особым показаниям. В редких случаях возможно развитие тендинитов (воспаление сухожилий, особенно ахилловых), удлинение интервала Q-T на электрокардиограмме (может провоцировать развитие желудочковых аритмий), фотодерматозы, возбуждение ЦНС (в редких случаях вызывают судороги, психозы, галлюцинации) (табл. 26.22).
Фторхинолоны не следует назначать больным с судорожным синдромом и выраженной цереброваскулярной недостаточностью. Изредка появляются артралгии, миалгии, аллергические кожные реакции (сыпь), лейкопения, эозинофилия, транзиторная гиперферментемия. Нефротоксический эффект не наблюдается.
Лекарственные взаимодействия
Хинолоны I поколения не следует сочетать с нитрофуранами, поскольку при этом резко снижается эффект; синергизм наблюдается при взаимодействии с хлорамфениколом, тетрациклинами, полиеновыми антибиотиками.
Всасывание фторхинолонов ухудшается при одновременном приеме антацидов, препаратов, содержащих кальций или алюминий, препаратов железа, сукральфата.
Таблица 26.22. Сравнительная оценка нежелательных лекарственных реакций фторхинолонов (в процентах)
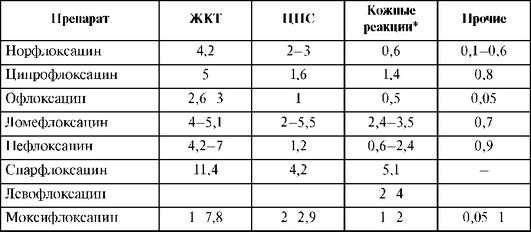
№ Включая фототоксичность. По сводным данным, охватывающая 1040 больных.
β-Лактамные антибиотики, аминогликозиды, цефалоспорины, макролиды можно применять в комбинации с хинолонами (целесообразно контролировать функции печени и назначить гепатопротекторы). Установлен синергизм пенициллинов и хинолонов в отношении синегнойной палочки.
Циметидин, ранитидин, метронидазол, клиндамицин, глибенкламид с ципрофлоксацином не взаимодействуют.
Фторхинолоны (особенно ципрофлоксацин, норфлокацинp и пефлоксацин) могут ингибировать метаболизм теофиллина в печени и повышать его концентрацию в крови.
При сочетании с НПВС возрастает риск нейротоксичности, вплоть до развития судорог.
Совместное назначение урикозурических препаратов (пробенецидx) приводит к замедлению выведения фторхинолонов и повышению его концентрации в плазме крови.
Рифампицин увеличивает клиренс ципрофлоксацина, их совместное применение может ослаблять антибактериальное действие, хотя эти положения требуют дальнейшего изучения (при туберкулезе не доказано).
Эффект варфарина при приеме ципрофлоксацина может усиливаться, но в меньшей степени, чем при приеме других хинолонов.
Ципрофлоксацин при сочетании с алкоголем снижает способность к концентрации внимания.
Важнейшие клинически значимые взаимодействия фторхинолонов представлены в табл. 26.23.
Таблица 26.23. Важнейшие взаимодействия фторхинолонов
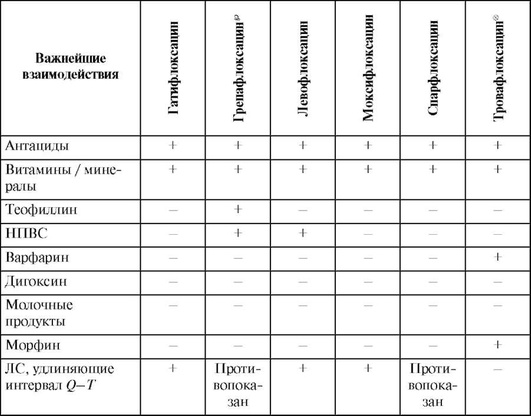
Показания
Противопоказания
Предостережения
Применение фторхинолонов у детей до 15-18 лет было ограничено ввиду возможного повреждения костно-суставной системы. В последнее время это ограничение оспаривается, хотя в фармакологических руководствах развитых стран подчеркивается, что безопасность ципрофлоксацина для лиц моложе 18 лет пока не доказана. Однако не исключается возможность их назначения по жизненным показаниям.
Ципрофлоксацин противопоказан лицам старческого возраста, а также страдающим эпилепсией и поражениями ЦНС. У пожилых пациентов увеличивается риск разрывов сухожилий, особенно при одновременном приеме глюкокортикоидов.
Нитрофураны
Нитрофураны оказывают преимущественно бактериостатический эффект, устойчивость к ним развивается медленно. Спектр антимикробного действия довольно широк: нитрофураны подавляют грамположительные (стрептококки, пенициллиназопродуцирующие стафилококки), грамотрицательные микроорганизмы (кишечные палочки, сальмонеллы, шигеллы, клебсиеллы, энтеробактеры) и многие простейшие (лямблии, трихомонады). Антимикробное действие не снижается в присутствии гноя и других продуктов тканевого распада. По спектру действия и фармакокинетическим характеристикам представители этой группы препаратов существенно отличаются друг от друга.
Клиническая фармакология нитрофуранов
Нитрофурал (Фурацилин*), в связи с производством более безопасных и эффективных нитрофуранов, применяется только местно (полоскание зева, промывание раневых поверхностей, при лечении гнойных ран), в настоящее время к нему устойчивы возбудители многих внутрибольничных инфекций.
Нитрофурантоин (Фурадонин*) хорошо всасывается, его применяют только при инфекциях мочевыводящих путей, что обусловлено особенностями его фармакокинетики: он чрезвычайно быстро выводится почками (T1/2 составляет 20-60 мин), поэтому в крови и тканях средняя терапевтическая концентрация не достигается. В щелочной моче антимикробный эффект усиливается. Нитрофурантоин существенно токсичнее других перорально назначаемых нитрофуранов. Вероятность токсических эффектов препарата повышается при нарушении выделительной функции почек (при снижении клиренса креатинина ниже 40 мл/мин препарат не назначают) и при сочетании с препаратами, подкисляющими мочу.
Фуралтадонx по спектру антимикробной активности мало отличается от фуразолидона, лучше всасывается, менее активен по отношению к простейшим. Средняя терапевтическая концентрация после приема разовой дозы удерживается в течение 4-6 ч. При кислой реакции мочи реабсорбция препарата усиливается, что может привести к кумуляции. При низких значениях рН и олигурии препарат отменяют.
Нифурател (Макмирор*) оказывает противопротозойное и противогрибковое действие. Обладает широким спектром антимикробного действия, активен в отношении Trichomonas vaginalis, высокоактивен в отношении рода Candida. Нифурател применяют для лечения вульвовагинальных инфекций, инфекционно-воспалительных заболеваний мочевыводящих путей, кишечного амебиаза и лямблиоза. Возможны аллергические реакции и диспепсические явления. Противопоказан при беременности и лактации.
Нифуроксазид - это противомикробное средство широкого спектра действия (грамположительные возбудители: стафилококки, стрептококки, гемофильная палочка; грамотрицательные микроорганизмы: сальмонеллы, энтеробактер; кишечная палочка, протей). Нифуроксазид не нарушает равновесия кишечной микрофлоры. Имеет низкую абсорбцию, поэтому его применяют при диарее инфекционного генеза. Нифуроксазид противопоказан при гиперчувствительности к препарату и нитрофуранам, а также у новорожденных.
Основная особенность фармакокинетики фуразидина (Фурагин*) - относительно медленное выведение, поэтому его можно назначать 2 раза в сутки; в моче наблюдается более низкая концентрация препарата, чем на фоне нитрофурантоина.
Растворимый фуразидин применяют внутривенно капельно, введение суточной дозы (0,1% водный раствор) обеспечивает среднюю терапевтическую концентрацию в крови и тканях в течение 48 ч.
Фуразолидон подавляет развитие трихомонад и лямблий, грамположительных и грамотрицательных микроорганизмов. Блокирует МАО. Фуразолидон назначают внутрь после еды. Этот препарат всасывается хуже, чем нитрофурантоин и фуразидин (Фурагин*), и его терапевтическая концентрация сохраняется в крови 4-6 ч. В терапевтических концентрациях фуразолидон обнаруживается в желчи и моче. Выделяясь с желчью, препарат оказывает благотворный эффект при кишечных инфекциях. Особенно фуразолидон активен по отношению к возбудителям дизентерии, брюшного тифа и паратифов, при лямблиозе. Основной путь элиминации этого препарата - почечная экскреция. При выраженных нарушениях клубочковой фильтрации кумулирует в крови.
Лекарственные взаимодействия
Опасно сочетание нитрофуранов с кислыми препаратами (аскорбиновая кислота, кальция хлорид, ЛС, содержащие аммония хлорид).
В связи со способностью фуразолидона ингибировать МАО его противопоказано сочетать с другими препаратами, ингибирующими этот фермент (ипрониазидx, ниаламидx и др.). Такая комбинация может вызвать артериальную гипертензию за счет увеличения активности эндогенных катехоламинов.
Нежелательные лекарственные реакции
Диапазон между терапевтическими и токсическими дозами нитрофуранов достаточно велик. Среди назначаемых внутрь препаратов наиболее токсичен нитрофурантоин (Фурадонин*); НЛР при использовании фуразолидона, фуразидина (Фурагин*) и фуралтадона8 встречаются реже и примерно с одинаковой частотой. У новорожденных и при почечной недостаточности вероятность токсических осложнений существенно возрастает.
Наиболее часто встречаются диспепсические расстройства (тошнота, рвота, боль в животе), особенно характерные для нитрофурантоина. Для профилактики диспепсических расстройств нитрофураны назначают после еды, запивая щелочными растворами. Значительно реже встречаются неврологические (моно- и полиневриты) и гематологические (гемолитическая и мегалобластная анемии) осложнения, поражение кожи (лейкодерма) и бронхолегочные проявления (отек легких, бронхоспазм, а также пневмонит у женщин старше 60 лет, сопровождающийся лихорадкой и эозинофилией). У новорожденных и грудных детей возможно образование метгемоглобина. Аллергические реакции при использовании нитрофуранов встречаются редко и в основном ограничиваются поражением кожи.
Специфическое свойство нитрофуранов, имеющее практическое значение, заключается в их способности снижать толерантность к алкоголю. Это антабусоподобный эффект, сохраняющийся в течение 5- 7 дней после отмены нитрофуранов.
8-Оксихинолины
Клиническая фармакология 8-оксихинолинов
Бактерицидное действие нитроксолина реализуется путем комплексирования их с ионами металлов, необходимых для активации ферментных систем микроорганизмов. Препарат действует бактерицидно, селективно ингибирует синтез бактериальной ДНК. Спектр действия включает грамотрицательные бактерии, амебы, грибы рода Candida. Нитроксолин хорошо всасывается в кишечнике. Он очень быстро выводится почками, что обусловливает низкую концентрацию в плазме крови (при сохраненных функциях почек). Показанием к назначению нитроксолина служат инфекции мочевыводящих путей, вызванные грамотрицательной микрофлорой. Нитроксолин можно сочетать с противогрибковыми антибиотиками и сульфаниламидами. Не применяют у новорожденных и у больных со сниженным функционированием почек.
Также используют для лечения дизентерии, сальмонеллеза, пищевых токсикоинфекциях, вызванных стафилококками и энтеробактериями, при энтероколите, дисбактериозе, вагините, трихомониазе.
Нитроимидазолы
Группа нитроимидазолов представлена синтетическими препаратами: метронидазолом, тинидазолом, орнидазолом.
Клиническая фармакология нитроимидазолов
Фармакодинамика
Нитроимидазолы избирательно воздействуют на микроорганизмы, ферментные системы которых способны восстанавливать нитрогруппу. Активные восстановленные формы препаратов нарушают репликацию ДНК и синтез белка в клетке микроорганизма, ингибируют тканевое дыхание.
Спектр активности
Препараты обладают высокой активностью в отношении анаэробных бактерий и простейших. К нитроимидазолам чувствительны простейшие (Trichomonas vaginalis, Entamoeba histolytica, Giardia lamblia, Lamblia intestinalis, Escherichia coli, Leishmania spp.), а также Helicobacter pylori.
Фармакокинетика
Нитроимидазолы хорошо всасываются при приеме внутрь. Биодоступность метронидазола более 80%, орнидазола - 90%, тинидазола - 100%, и этот показатель не зависит от приема пищи. После ректального введения метронидазола в свечах его биодоступность ниже на 10%, чем при приеме внутрь.
Метаболизм
Нитроимидазолы метаболизируются в печени с образованием активных и неактивных метаболитов (30-60% метронидазола превращается в активный метаболит 2-оксиметронидазол, который оказывает противопротозойное и антимикробное действие).
Распределение
Нитроимидазолы хорошо распределяются, легко проникает в ткани, абсцессы, проходят через ГЭБ, создавая высокие концентрации в ликворе и в ткани мозга (метронидазол), в плаценту; попадают в грудное молоко, выделяются со слюной и желудочным соком.
Выведение T1/2 метронидазола составляет 8-12 ч, при алкогольном поражении печени - 10-29 ч. Препарат выводится из организма почками - 60-80% от принятой дозы, примерно 20% - в неизмененном виде; экскреция с желчью - 50%, где его концентрация может превышать концентрацию в крови, затем в кишечнике метронидазол вновь всасывается. При дефекации выводится до 6-15%. При повторных введениях возможна кумуляция. При выраженной почечной недостаточности (клиренс креатинина <10 мл/мин) суточную дозу необходимо уменьшить вдвое. Метронидазол и его основные метаболиты быстро удаляются из крови при гемодиализе (T1/2 сокращается до 2,6 ч).
T1/2 тинидазола составляет 12-14 ч. Препарат выделяется с грудным молоком в течение 72 ч после приема. Экскреция с желчью составляет 50%, почками - 25%, в неизмененном виде - 12%, остальное количество - с калом.
T1/2 орнидазола составляет около 13 ч. Экскретируется в виде метаболитов с мочой (60-70%), калом (20-25%). Около 5% выводится в неизмененном виде.
Нежелательные лекарственные реакции
НЛР представлены в табл. 26.24.
Таблица 26.24. Нежелательные лекарственные реакции, вызванные нитроимидазолами
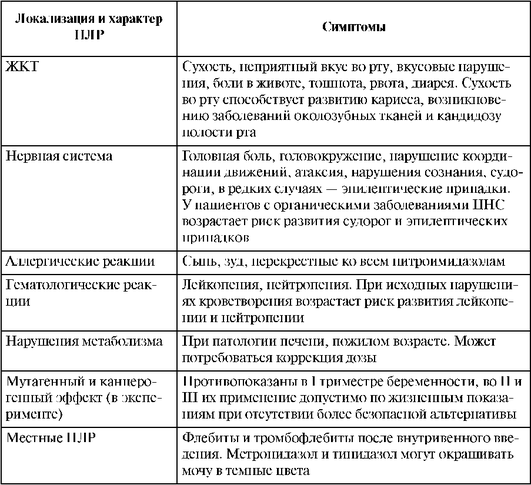
Показания к применению
Противопоказания
Применение нитроимидазолов в период грудного вскармливания не рекомендуется, поскольку концентрация метронидазола и его метаболитов в плазме крови ребенка составляет 10-20% от концентрации в крови матери.
Лекарственные взаимодействия
Лекарственные взаимодействия нитроимидазолов приведены в табл. 26.25.
Таблица 26.25. Лекарственные взаимодействия нитроимидазолов
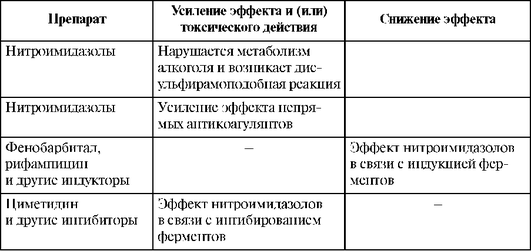
Чувствительность и резистентность микроорганизмов к антибактериальным лекарственным средствам
Чувствительность возбудителя характеризуется минимальной концентрацией антибиотика в питательной среде, подавляющей рост микроорганизмов. Оценка противомикробной активности проводится in vitro.
Минимальная подавляющая концентрация - это наименьшая концентрация АМП, которая через определенный период времени приостанавливает рост специфических микроорганизмов in vitro. Результаты определения чувствительности анализируют по отдельным видам микроорганизмов и их штаммов. Данные представляют в виде концентрации препарата, необходимой для подавления 50% штаммов (МПК50) или для подавления 90% штаммов (МПК90).
Минимальная бактерицидная концентрация - это наименьшая концентрация АМП, вызывающая гибель посеянных на питательную среду микроорганизмов через определенный период времени.
Постантибиотический эффект - это промежуток времени между уменьшением концентрации препарата ниже значения МПК и возобновлением логарифмического роста популяции микроорганизмов. Постантибиотический эффект рассчитывают как разницу между временем, необходимым микроорганизмам, подвергшимся и не подвергшимся (контрольная группа) действию лекарственного вещества, чтобы их количество увеличилось в 10 раз по сравнению с количеством сразу после отмены ЛС. Постантибиотический эффект оценивают в минутах и часах.
По степени чувствительности к антибактериальным препаратам бактерии разделяют:
Чувствительными считаются микроорганизмы, рост и размножение которых в очагах инфекции (крови) прекращается при средних терапевтических концентрациях лекарства в сыворотке крови (после назначения обычных доз).
Умеренно чувствительными считаются микроорганизмы, для угнетение роста которых требуются максимальные дозы лекарственного препарата.
Устойчивыми (резистентными) считаются микроорганизмы, бактериостатический эффект по отношению к которым может быть достигнут только in vitro при высоких концентрациях лекарственного препарата, токсичных для человека.
Резистентность - это появление субпопуляции микроорганизмов, для которых МПК выше, чем для исходного штамма. Антибиотикорезистентность представляет собой неизбежное биологическое явление, предотвратить которое практически невозможно.
Антибиотикорезистентные микроорганизмы опасны не только для пациента, у которого они были выделены, но и для многих других людей, даже разделенных временем и пространством, поэтому борьба с антибиотикорезистентностью приобрела глобальные масштабы. Клиническое значение сниженной чувствительности микроорганизмов in vitro зависит от того, превышает ли МПК для этой субпопуляции те концентрации АМП, которые могут быть достигнуты в крови, тканях или биологических жидкостях при назначении обычных доз ЛС.
Виды резистентности:
-
природная резистентность - это генетически обусловленное отсутствие чувствительности микроорганизма к противомикробным средствам (например, устойчивость вирусов к антибиотикам, грамотрицательных бактерий к бензилпенициллину, анаэробных бактерий к цефалоспоринам I поколения и др.);
-
приобретенная резистентность - это устойчивость, возникающая в результате мутации отдельных штаммов бактерий и селекции устойчивых клонов микроорганизмов или в результате внехромосомного (плазмидного) обмена генетической информацией между отдельными бактериальными клетками.
Выделяют два типа приобретенной резистентности бактерий:
-
первичная резистентность - это устойчивость микроорганизма до начала лечения. Она определяет выбор антибактериального препарата для лечения (например, первичная устойчивость некоторых штаммов пневмококка или золотистого стафилококка к бензилпенициллину делает бессмысленным его назначение);
-
вторичная резистентность бактерий возникает или возрастает в процессе лечения антибактериальными препаратами и требует пересмотра тактики лечения.
Резистентность микроорганизмов носит строго специфический характер в отношении отдельных антибактериальных препаратов или нескольких ЛС в пределах одной группы. Полная перекрестная резистентность наблюдается среди микроорганизмов, устойчивых к природным тетрациклинам, частичная - среди микроорганизмов, устойчивых к макролидам.
Механизмы развития бактериальной резистентности к противомикробным средствам
Резистентность может развиваться у большинства микроорганизмов. Механизмы ее реализации разнообразны.
I механизм
Наиболее распространенный механизм защиты бактерий от действия антибиотиков заключается в образовании ферментов - β-лактамаз, нарушающих целостность β-лактамного кольца, тем самым инактивируя антибиотики. β-Лактамазы часто вырабатываются такими микроорганизмами, как стафилококки, кишечная палочка, гонококки, анаэробы. β-Лактамазы классифицируют по субстратному профилю (пенициллиназы, цефалоспориназы, карбапенемазы и др.) и по локализации генов в клетке микроорганизма, кодирующих продукцию р-лактамаз (хромосомные и плазмидные).
II механизм
II механизм обусловлен изменением участка клетки микроорганизма, на которую действует антибиотик (модификация мишени), и антибиотик не может связаться с мишенью, на которую направлена его активность. Пример таких микроорганизмов - пенициллинрезистентный пневмококк.
III механизм
III механизм обусловлен изменением клеточных структур-мишеней для антибиотиков: синтез микроорганизмов нового дополнительного пенициллинсвязывающего белка - механизм «обходного пути». При отсутствии у микроорганизмов этого механизма β-лактамные антибиотики, связываясь с пенициллинсвязывающими белками, блокируют транспептидазы и карбоксипептидазы микроорганизмов, отвечающие за синтез пептидогликанов клеточной стенки, нарушая тем самым синтез клеточной стенки бактерии (механизм действия β-лактамов). Вследствие этого механизма резистентности, несмотря на действие антибиотика, жизнедеятельность микроорганизма не нарушается. Такой механизм характерен для стафилококков, устойчивых к метициллину, оксациллину и другим пенициллинам и цефалоспоринам. Ингибиторы β-лактамаз в этих случаях неэффективны.
VI механизм
VI механизм обусловлен нарушением проницаемости клеточной стенки для β-лактамов. Такой механизм служит причиной устойчивости синегнойной палочки к р-лактамам.
V механизм
V механизм обусловлен активным выведением антибактериальных препаратов из клетки микроорганизма (эффлюкс). В этом случае β-лактамазы, продуцируемые грамположительными микроорганизмами, выделяются из клетки в межклеточное пространство, а продуцируемые грамотрицательными бактериями не покидают клетку и циркулируют между наружной и внутренней мембраной. В настоящее время выделено более 200 видов β-лактамаз, классифицированных по субстратному профилю (пенициллиназы, цефалоспориназы, карбапенемазы), по локализации в клетке микроорганизма генов, кодирующих продукцию β-лактамаз (хромосомные и плазмидные). Наиболее распространенные β-лактамазы и их свойства представлены в табл. 26.26.
Таблица 26.26. Наиболее распространенные β-лактамазы и их свойства
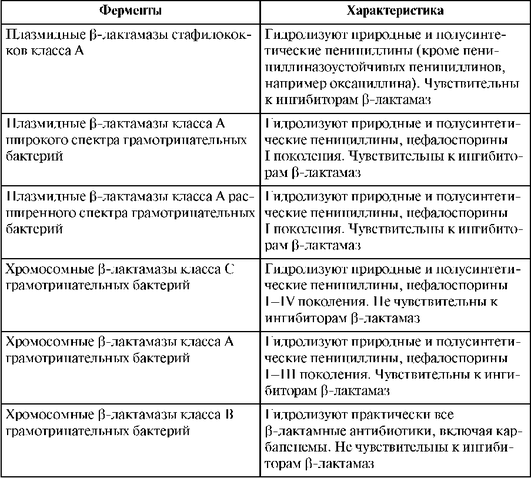
Активность антибиотиков в отношении микроорганизмов, вырабатывающих β-лактамазы, различна и зависит от типа β-лактамаз, вырабатываемых микроорганизмом (табл. 26.27).
Таблица 26.27. Влияние β-лактамаз на активность антибиотиков
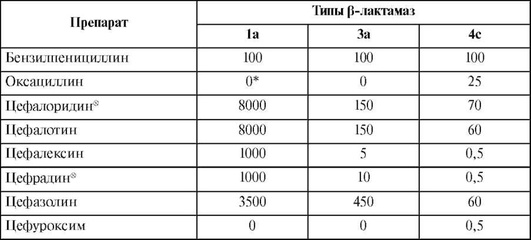
* 0 - отсутствие влияния.
β-Лактамазы типа 1а разрушают преимущественно цефалоспорины, типа 3а - цефалоспорины и пенициллины, но не действуют на оксациллин, типа 4с - разрушают цефалоспорины и все пенициллины (в том числе и пенициллиназоустойчивые).
На протяжении последних 10 лет во всем мире отмечается значительный рост устойчивости возбудителей внебольничных и нозокомиальных инфекций. При этом следует принимать во внимание уровень проблемы: региональный или локальный (стационар, отделение). Прежде всего, необходимо учитывать глобальные тенденции в развитии антибиотикорезистентности. Например, появление и широкое распространение метициллинрезистентных стафилококков, пенициллинрезистентных пневмококков, устойчивость у клебсиелл вследствие появления β-лактамаз расширенного спектра действия. При всей важности проблемы глобальных тенденций, при планировании политики антибиотико-терапии более логично опираться на результаты эпидемиологических исследований в конкретной стране, городе (регионе) или локальные исследования (особенно при лечении нозокомиальных инфекций).
β-Лактамазы расширенного спектра действия способны разрушать пенициллины, цефалоспорины I-IV поколения, монобактамы; AmpC β-лактамазы разрушают пенициллины, цефалоспорины I-III поколения, монобактамы. Из всех β-лактамов только карбапенемы отличаются высокой стабильностью к гидролизу всеми хромосомными и плазмидными β-лактамазами, в том числе β-лактамазами расширенного спектра действия.
Для преодоления резистентности, связанной с продукцией β-лактамаз, были разработаны соединения, способные подавлять активность этих ферментов, - так называемые ингибиторы β-лактамаз: клавулановая кислота (клавуланат), сульбактам и тазобактам. Они используются при создании комбинированных (ингибиторзащищенных) пенициллинов (амоксициллин + клавулановая кислота, ампициллин + сульбактам и др.) и цефалоспоринов (цефоперазон + сульбактам).
В последнее десятилетие серьезную проблему представляют нозокомиальные (внутрибольничные) инфекции, и здесь отчетливо прослеживаются две тенденции.
Первая тенденция современной антибиотикотерапии связана с изменением спектра возбудителей госпитальных инфекций, прежде всего в отделениях интенсивной терапии и реанимации, за счет увеличения частоты грамположительных микроорганизмов, вызывающих тяжелые формы инфекции (пневмонии, раневая инфекция, сепсис).
Вторая тенденция связана с существенным повышением резистентности грамотрицательных микроорганизмов к цефалоспоринам III поколения, в основном за счет образования ими β-лактамаз различных типов и классов. Резистентность обусловлена гиперпродукцией хромосомных β-лактамаз (вследствие мутаций регуляторных областей генома, усиливающих синтез фермента). Этот механизм обусловливает достаточно высокую частоту возникновения вторичной резистентности в процессе лечения цефалоспоринами III поколения пациентов с тяжелыми госпитальными пневмониями или сепсисом, вызванными Enterobacter spp. и Serratia marcescens (селекция мутантов-гиперпродуцентов на фоне элиминации чувствительных микроорганизмов). В настоящее время частота штаммов гиперпродуцентов хромосомных β-лактамаз Enterobacter spp., резистентных к цефалоспоринам III поколения, в большинстве клиник Европы колеблется от 30 до 60%.
Второй важный механизм развития резистентности микроорганизмов к цефалоспоринам заключается в образовании плазмидных β-лактамаз расширенного спектра (наиболее часто происходит у штаммов Klebsiella spp. - примерно в 30%), гидролизующих все цефалоспорины III поколения.
Рутинные лабораторные методы оценки антибиотикочувствительности часто не определяют резистентность по этому механизму, и лаборатория может выдать врачу неправильный результат, что создает дополнительные сложности лечения таких инфекций.
Указанные выше две тенденции стимулируют поиск новых антибактериальных препаратов, которые, с одной стороны, позволили бы преодолеть проблему мультирезистентности грамотрицательных возбудителей госпитальных инфекций, особенно продуцентов β-лактамаз расширенного спектра, а с другой стороны, обладали бы более высокой активностью в отношении грамположительных микроорганизмов.
Поиск таких препаратов привел к получению в середине 1990-х годов соединений, разрушающих β-лактамазы (сульбактам, тазобактам, клавулановая кислота), а также к созданию на их основе комбинированных препаратов (амоксициллин + клавулановая кислота, пиперациллин + тазобактам, тикарциллин + клавулановая кислота, ампициллин + сульбактам, цефоперазон + сульбактам) и двух новых цефалоспориновых антибиотиков, отнесенных к цефалоспоринам IV поколения, - цефепима и цефпирома, характеризующихся широким, хорошо сбалансированным противомикробным спектром.
Пневмококк не продуцирует β-лактамаз, и в этом случае резистентность к лечению антибактериальными ЛС возникает вследствие модификации пенициллинсвязывающих белков на поверхности клеточной стенки. Для преодоления такой резистентности использовать ингибиторы β-лактамаз бессмысленно.
В последние годы в ряде стран отмечается распространение пенициллинустойчивых пневмококков, а также штаммов, устойчивых к макролидным антибиотикам, тетрациклину и ко-тримоксазолу. В некоторых регионах устойчивость к макролидам превышает устойчивость к пенициллину. Наличие у пневмококка пенициллинрезистентности часто сопровождается мультирезистентностью в отношении других р-лактамных антибиотиков.
Устойчивость Streptococcus pneumoniae к пенициллинам сильно варьирует в зависимости от региона: в Венгрии наблюдается до 50% резистентных штаммов (1992), во Франции - 25-35% (1990), в Великобритании к пенициллинам устойчивы только 3-4% микроорганизмов.
Распространенность пенициллинрезистентного пневмококка в России пока низкая: от 7,5% (дети) до 9% (взрослые). Максимальный уровень резистентности к пенициллинам наблюдается в Москве и крупных городах. Однако штаммы с высоким уровнем устойчивости (МПК90 >0,2 мкг/мл) в России не выявлены, а все микроорганизмы с промежуточной резистентностью сохраняли устойчивость к цефалоспоринам и амоксициллину. Около 65% штаммов пневмококков резистентны к тетрациклину, а 62% - к ко-тримоксазолу.
В условиях нашей страны препаратами выбора при лечении инфекций, вызываемых Streptococcus pneumoniae, служат аминопенициллины. При выделении пневмококка с МПК пенициллинов более 2,0 мг/л рекомендуется применять цефтриаксон или цефотаксим.
Методы диагностики и контроля
Методы бактериологического контроля:
При наличии субстрата для исследования забор материала для бактериологических исследований должны по возможности проводить до начала лечения АМП, что значительно повышает информативность и достоверность исследований. Достоверность исследований зависит также от правильности забора и транспортировки материала для исследования. В процессе антибиотикотерапии необходим постоянный микробиологический мониторинг.
Методы иммунологического контроля: культуральная диагностика внутриклеточных возбудителей доступна только специализированным лабораториям, поэтому общепринятым методом служит серотипирование.
Используют реакции непрямой иммунофлюоресценции и связывания комплемента. Для всех возбудителей абсолютно доказано 4-кратное увеличение титров антител в парных сыворотках крови, взятых с интервалом в 2 нед. Эти методы предоставляют лишь ретроспективную информацию. С недавних пор для диагностики микоплазменной инфекции исследуют сыворотку крови на наличие специфических антител к Mycoplasma pneumoniae классов IgM и IgG иммуноферментным методом ELISA. Повышенные концентрации антител класса IgM свидетельствуют об острой фазе инфекционного процесса, после которого повышаются концентрации антител класса IgG, способные сохраняться в течение длительного времени. Этот метод более чувствителен, чем реакции связывания комплемента и непрямой иммунофлюоресценции, и не требует изучения парных сывороток. (Тест ELISA также можно применять для обнаружения антигена микоплазмы в мокроте.) Для определения концентрации специфических антител к хламидиям в сыворотке крови, кроме реакций связывания комплемента и непрямой иммунофлюоресценции, также можно применять тест ELISA. Для верификации альтернативных возбудителей используются и методы, основанные на ПЦР.
Общие принципы фармакотерапии инфекционно-воспалительных заболеваний
Антимикробные ЛС необходимо назначать при воспалительных заболеваниях, в этиологии которых ведущую роль играет чувствительная к ним бактериальная микрофлора, при бактериальном носительстве и для периоперационной профилактики. В каждом случае надо определить показания к их применению.
При лечении неосложненных острых вирусных заболеваний дыхательных путей антибиотики применяться не должны, поскольку они не оказывают противовирусного действия и не предотвращают развитие бактериальных осложнений.
Антимикробная терапия может быть этиотропной и эмпирической.
Этиотропная терапия более рациональна и предполагает целенаправленное применение АМП препаратов против установленного возбудителя инфекционного процесса.
Эмпирическая, или стартовая, терапия - это применение АМП до получения сведений о возбудителе и его чувствительности к тем или иным препаратам.
Эмпирическую терапию проводят с учетом наиболее вероятных возбудителей данной инфекции и наиболее вероятной их чувствительности к доступным АМП. По возможности следует учитывать локальные результаты эпидемиологических исследований антибиотикорезистентности возбудителей в регионе, популяции, в лечебном учреждении.
Для повышения эффективности лечения бактериальных инфекций необходимы:
-
точный диагноз, позволяющий определить наличие общего или локального воспаления, вызванного бактериальным возбудителем;
-
определение предполагаемого возбудителя и обоснование применения тех или иных антибактериальных препаратов;
-
забор материала для бактериологического и иммунологического исследования (при наличии показаний - взятие субстрата для исследования в бактериологической лаборатории);
-
выбор оптимального препарата в соответствии с инфекционно-воспалительным процессом, его локализацией и тяжестью;
-
выбор оптимальной дозы (с учетом правил дозирования отдельных препаратов), кратности (исходя из функционирования почек или печени) и пути введения (с учетом тяжести состояния больного);
-
учет особенностей фармакокинетики препаратов (биодоступность, распределение, проникновение через физиологические барьеры, концентрация в жидкостях и тканях организма, метаболизм, скорость выведения);
-
использование препаратов с узким спектром действия для эмпирической терапии нетяжелых заболеваний или заболеваний средней тяжести, а при тяжелом течении - либо использование препаратов более широкого спектра действия, либо комбинированной терапии;
-
быстрая смена антибиотиков и коррекция схем лечения при получении новых данных о возбудителе (если отсутствует эффект от эмпирической терапии);
-
выбор методов контроля за эффективностью и безопасностью терапии.
Врачу при назначении антибиотикотерапии необходимо учитывать особенности пациента: возраст, массу тела, сопутствующие заболевания, функциональное состояние печени и почек, прием других, особенно жизненно важных ЛС, беременность, кормление грудью и тому подобное, а также фармакологический анамнез о применяемых ранее АМП, их эффективности и переносимости.
Этиологическое лечение - это целенаправленный выбор АМП или их комбинации в соответствии с видом возбудителя (или возбудителей), выделенного из патологического материала при бактериологическом исследовании или установленного другими методами (иммуноферментный, ПЦР и др.)
Целенаправленный выбор противомикробных ЛС основывается на грамотной идентификации микроорганизмов, вызвавших инфекционное заболевание, и определении чувствительности микроорганизмов к АМП (грамотная бактериологическая и ПЦР-диагностика).
При выделении «проблемных возбудителей»: стафилококков, кишечной палочки, клебсиелл, энтерококков, протея, синегнойной палочки, микобактерий туберкулеза - необходимо в обязательном порядке определять их чувствительность к АМП.
Целенаправленный выбор антибиотика чаще осуществляют при необходимости смены ЛС в случае неэффективности лечения стартовым препаратом (лекарственным препаратом первого выбора). При тяжелом течении заболевания, нозокомиальных инфекциях, выделении «проблемных» возбудителей в выборе лекарственного препарата необходимо учитывать антибиотикорезистентность. В этих случаях для контроля за возможным развитием суперинфекции (смена возбудителя, возникновение ассоциации микроорганизмов и т.д.) и развитием вторичной резистентности показан также микробиологический мониторинг, представляющий собой один из методов оценки динамики заболевания и контроля за эррадикацией возбудителя.
Если клиническая картина заболевания характерна для определенного возбудителя, для которого нетипично развитие резистентности, то лечение можно проводить без определения чувствительности микроорганизма к антибиотикам. Например, возбудителями рожистого воспаления и скарлатины всегда бывают стрептококки, эпидемического менингита - менингококки. Однако стремительное развитие резистентности микрофлоры все реже позволяет обходиться без бактериологического контроля, особенно при тяжелом течении заболевания.
При проведение антимикробной терапии важно:
-
знать особенности фармакокинетики препаратов (биодоступность, распределение, проникновение через физиологические барьеры, концентрация в жидкостях и тканях организма, метаболизм, скорость выведения);
-
знать и учитывать особенности больного (возраст, масса тела, анамнез, сопутствующая патология, функции печени и почек, прием других ЛС, беременность или кормление грудью);
-
выбирать оптимальный режим дозирования препарата (с учетом правил дозирования отдельных препаратов), кратности (исходя из функций почек или печени) и пути введения (с учетом тяжести состояния);
-
замену АМП при выработке у микроорганизма к нему вторичной устойчивости проводят с учетом перекрестной устойчивости.
С позиций фармакотерапии и клинической фармакологии важно определение программ контроля эффективности антибиотикотерапии, а также программ контроля прогнозируемых НЛР.
Вопросы о предполагаемой длительности лечения, о микробиологическом и лекарственном мониторинге и, наконец, об оптимальной стоимости лечения представляют большой интерес.
При противомикробной терапии выбор препарата и режима дозирования осуществляют на основе знаний клинической фармакологии ЛС.
Фармакодинамика антимикробных лекарственных средств
В широком смысле под фармакодинамикой понимают действие ЛС на специфические рецепторы живого организма (механизм действия) и возникающие в результате этого эффекты. Поскольку организм человека не является (или не должен являться) мишенью действия антимикробных ЛС, фармакодинамика в применении к ним - это действие на микроорганизм. Таким образом, фармакодинамической характеристикой антибиотика служат спектр и степень его активности в отношении того или иного вида микроорганизмов. Количественное выражение активности АМП - его МПК. Чем она меньше, тем он более активен. Нередко в аннотациях к АМП приводится большой список микроорганизмов, к которым показана активность in vitro, однако реальное значение имеет только то, по отношению к каким возбудителям инфекций доказана эффективность терапии не только в ходе микробиологических исследований, но и подтверждена клинической практикой. Поэтому микробиологическая активность препарата in vitro - это только первая предпосылка для обеспечения клинической и микробиологической эффективности.
В последние годы трактовка фармакодинамики АМП расширилась. В нее входит взаимоотношение между концентрациями препарата в организме или в искусственной модели и его антимикробной активностью. Исходя из этого выделяют две группы антибиотиков: с концентрационнозависимой антимикробной активностью и с времязависимой активностью. Для первой группы препаратов, примером которых могут служить аминогликозиды или фторхинолоны, степень гибели бактерий коррелирует с концентрацией антибиотика в биологической среде, например в сыворотке крови. Поэтому цель режима дозирования заключается в достижении максимально переносимой концентрации препарата.
Для АМП с времязависимым антимикробным действием наиболее важное условие - это длительное поддержание концентрации на относительно невысоком уровне (в 3-4 раза выше МПК), причем при повышении концентрации препарата эффективность лечения не возрастает. К антимикробным препаратам с времязависимым типом действия относятся пенициллины, цефалоспорины. Цель режимов их дозирования заключается в поддержании в сыворотке крови и очаге инфекции концентрации препарата, в 4 раза превышающей МПК, в течение всего интервала между введением препарата, и при этом достаточно, чтобы такая концентрация сохранялась в течение 40-60% временного интервала между дозами.
К фармакодинамическим характеристикам антимикробных ЛС относят также типы (эффекты) их действия - бактерицидный (вызывает гибель инфекционного агента) и бактериостатический (приостанавливает размножение микроорганизма).
Термины «бактериостатический» и «бактерицидный» относительны, так как одни и те же ЛС могут обладать и тем и другим действием, что определяется видом микроорганизма, концентрацией АМП и длительностью экспозиции. Так, ванкомицин в отношении стрептококков и энтерококков оказывает бактериостатическое действие, а в отношении стафилококков - бактерицидное действие. Макролиды обычно действуют бактериостатически, однако в высоких концентрациях (в 2-4 раза превышающие МПК) они оказывают бактерицидный эффект на Streptococcus pyogenes, S. pneumoniae. Выделение бактерицидных и бактериостатических АМП имеет практическое значение при лечении тяжелых инфекций, особенно у пациентов с нарушениями иммунитета. Это связано с тем, что при нормальном иммунитете приостановление размножения микроорганизмов оказывается вполне достаточным, чтобы элиминацию патогенных микроорганизмов завершила иммунная система.
Выделение бактерицидных и бактериостатических АМП имеет практическое значение при лечении тяжелых инфекций, особенно у пациентов с нарушениями иммунитета. Это связано с тем, что при нормальном иммунитете приостановление размножения микроорганизмов оказывается вполне достаточным, чтобы элиминацию патогенных микроорганизмов завершила иммунная система.
Бактерицидные препараты служат препаратами выбора при тяжелых инфекциях или у пациентов с нарушениями иммунитета: у больных СПИДом, лиц, получающих иммуносупрессивную терапию или цитостатики, больных бактериальным эндокардитом, остеомиелитом, менингитом, нейтропенической лихорадкой, при тяжелых инфекциях головы и шеи (эндофтальмит, ангина Людвига) и др.
Количество антибиотиков-бактериостатиков невелико: тетрациклины, сульфаниламиды, хлорамфеникол, природные макролиды, налидиксовая кислота и некоторые другие. Вновь создаваемые АМП, поступающие на фармацевтический рынок, обладают преимущественно бактерицидным эффектом действия.
Фармакокинетика противомикробных лекарственных средств
С фармакокинетической точки зрения целью противомикробной химиотерапии является создание и поддержание в очаге бактериальной инфекции на протяжении всего периода лечения концентрации активного препарата, обеспечивающей бактерицидный (для бактериолитиков) или бактериостатический (для бактериостатиков) эффект в отношении предполагаемого или установленного возбудителя (возбудителей).
Возможность достижения этой концентрации зависит от физико-химических свойств ЛС, всасывания, метаболизма, связи с белками, экскреции, пути введения и режима дозирования препаратов. При выборе антибиотика и режима дозирования из фармакокинетических параметров наиболее важны T1/2, связь с белками, метаболизм, экскреция, а при приеме внутрь еще процент, стабильность и скорость всасывания, которые в основном определяют биодоступность ЛС.
При выборе ЛС также важны особенности распределения в органы и ткани, способность создавать в отдельных органах концентрации, равные или превышающие концентрацию в сыворотке крови.
При распространении бактериального воспаления за тканевые барьеры выбирают ЛС, способные проникать через тканевые барьеры, включая ГЭБ, и создавать там необходимый уровень концентрации (в плевральной, перитонеальной и суставной полости). На тканевое распределение наибольшее влияние оказывают связь с белками и липофильность. Вещества с высокой липофильностью легче проникают в ткани.
Распределяясь в организме, АМП связываются с белками плазмы крови. Высокое сродство с белками плазмы препятствует образованию высоких концентраций препарата во внесосудистом русле. Связывание антимикробных ЛС с белками не ограничивается сывороткой крови, происходит также в интерстициальной жидкости, в воспалительном экссудате и внутриклеточно (связывание с белками субклеточных структур, хроматином лейкоцитов и другими клеточными компонентами).
Также большое значение имеет степень ионизации антибиотика. Неионизированные соединения (например, макролиды) лучше проникают через липидные мембраны, в то время как для хорошего поступления в ткани легкого и накопления ЛС с высокой степенью ионизации (аминогликозиды и р-лактамные антибиотики) требуется кислая среда.
На концентрацию препарата в сыворотке крови влияет функциональное состояние печени и почек. Нарушение их функций требует замены препарата на другой, характеризующийся меньшим риском развития органотоксического эффекта. Если возможности такой замены при нарушении выделительной функции почек отсутствуют, необходимо обязательно изменить режим дозирования препарата с учетом степени нарушения выделительной функции почек по уровню креатинина или клиренса эндогенного креатинина.
Путь введения и режим дозирования противомикробных лекарственных средств
Для получения Cmax АМП в плазме крови после приема внутрь целесообразно принимать его натощак (пища замедляет абсорбцию большинства антимикробных ЛС). После еды назначают препараты, оказывающие местное раздражающее действие. Различают:
-
хорошо всасывающие антибактериальные средства (более 70%): хлорамфеникол, ампициллин, амоксициллин, доксициклин, рифампицин, цефалексин, цефуроксим, цефаклор, цефиксим, цефтибутен (Цедекс*), фузидовая кислота, новобиоцинx, фторхинолоны. Прием пищи минимально влияет на их биодоступность;
-
умеренно всасывающиеся (30-50%): феноксиметилпенициллин, оксациллин, эритромицин, тетрациклин, окситетрациклин, линкомицин;
-
плохо всасывающиеся (менее 30%): цефалоспорины (кроме указанных выше), бензилпенициллин, стрептомицин, ванкомицин, бацитрацин, аминогликозиды, полимиксины, нистатин, леворин.
Препараты с такой фармакокинетикой для системного применения применяют парентерально. Некоторые из них имеют пероральные формы (неомицин, полимиксин Мx) для лечения инфекций ЖКТ, где за счет плохого всасывания создают высокие концентрации ЛС.
Плохо всасывающиеся или раздражающие слизистую оболочку ЖКТ препараты вводят внутривенно или внутримышечно либо используют при лечении кишечных инфекций (неомицин). Необходимо учитывать, что в случае нарушений периферического кровообращения (токсикоз, недостаточность кровообращения) при внутримышечном введении их всасывание может замедляться, и тогда химиопрепараты также вводят внутривенно (желательно капельно, во избежание быстрого снижения их концентрации и местного раздражающего действия на стенку вен). При тяжелом течении инфекции сочетают первоначальное внутривенное струйное введение (возможно ни для всех антибиотиков) с последующим капельным или внутримышечным их назначением.
При распространении бактериального воспаления за тканевые барьеры (плеврит, перитонит, флегмона и т.д.) системное применение даже максимально допустимых доз антибиотиков не всегда позволяет получить эффективную концентрацию препаратов в очаге бактериальной инфекции. В таких случаях применяют комбинацию путей введения препарата: системное применение и введение непосредственно в полость. Более эффективны в этой ситуации методы непрерывной перфузии антибиотиков - диализ, которые позволяют постоянно поддерживать в очаге воспаления высокий уровень концентрации АМП, одновременно удаляя продукты воспаления и снижая риск развития НЛР, вызванных АМП.
Распределение противомикробных лекарственных средств
Большая часть антибиотиков хорошо распределяется в паренхиматозные органы, мягкие ткани (иначе они не внедряются в клиническую практику). Несколько хуже их распределение в миокард, костную ткань, поджелудочную железу, слезный мешок, предстательную железу. Многие антибиотики плохо проникают через неповрежденный ГЭБ. Большая часть антибиотиков хуже проходит через тканевые барьеры (в плевральную полость, перитонеальную полость, в полость суставов и т.д.), чем в паренхиматозные органы, поэтому тяжелое течение инфекционного воспаления этих локализаций требует применения высоких доз антибиотиков, что увеличивает риск развития НЛР и не всегда позволяет получить в очаге воспаления необходимый уровень концентрации антибиотика. При воспалении проницаемость тканевых барьеров значительно возрастает.
Распределение в органы, как правило, выражают в процентах к концентрации в сыворотке крови (или мкг/г ткани). Эта информация чаще представлена для новых антибиотиков. Для старых, а зачастую и для новых антибиотиков распределение оценивают в терминах «хорошее» (близко к концентрации в сыворотке крови), «удовлетворительное» (приблизительно 50-70% от концентрации в сыворотке крови), «плохое» (меньше 25% от концентрации в сыворотке крови).
Выбор препарата при равнозначном противомикробном спектре препаратов во многом обусловливают особенности распределения антибиотика в органы.
Метаболизм антимикробных лекарственных средств (общие вопросы)
Метаболизм антимикробных ЛС осуществляется преимущественно в печени. Метаболизирующиеся в этом органе лекарственные вещества подразделяют на две группы: препараты с высоким печеночным клиренсом и препараты с низким печеночным клиренсом. Для ЛС первой подгруппы характерна высокая степень извлечения (экстракции) из крови, что зависит от активности (емкости) метаболизирующих их ферментных систем. Поскольку ЛС этой группы быстро и легко метаболизируются в печени, печеночный клиренс определяется величиной и скоростью кровотока. Емкость ферментных систем для второй подгруппы ЛС относительно невелика, и в результате их печеночный клиренс зависит не от скорости печеночного кровотока, а от активности ферментов и степени связывания с белками крови.
Так, сульфонамиды в реакциях II фазы биотрансформации метаболизируются путем конъюгации с двумя эндогенными соединениями: глюкуронирование с эндогенной УДФ-глюкуроновой кислотой с участием фермента УДФ-глюкуронилтрансферазы (микросомы) и ацетилирование с ацетил-КоА с участием N-ацетил-трансферазы (цитозоль).
Взаимодействие АМП с другими ЛС происходит на уровне микросомального окисления.
Важным является вопрос о влиянии антибиотиков на лекарственные взаимодействия, опосредованные через цитохром Р450. Некоторые антибиотики клинически значимо могут изменять метаболизм других ЛС, что требует коррекции доз этих препаратов или даже замены одного из препаратов комбинации.
Известно, что многие ксенобиотики, в том числе и ЛС, ингибируя или индуцируя микросомальное окисление, могут существенно изменять метаболизм других ЛС при комбинированной терапии. Индукции могут подвергаться ферменты и I, и II фазы метаболизма. Это может приводить к повышению концентрации одного из компонентов комбинации (при ингибировании ферментов) или к снижению (при индукции микросомальных ферментов). Возможна самоиндукция (аутоиндукция), когда непосредственное воздействие молекул индуктора на регуляторную часть гена приводит к индукции фермента, увеличивая его активность и метаболизм ЛС [рифампицин, фенилбутазон (Бутадион*)].
Эритромицин, снижая активность ферментов, удлиняет T1/2 теофиллина, усиливает действие циклоспорина, карбамазепина, глюкокортикоидов, дигоксина, варфарина, эрготамина и др.
Совместное применение эритромицина и эрготамина может приводить к резким спастическим сосудистым реакциям. Эритромицин увеличивает AUC мидозаламаp (препарат из группы бензодиазепинов).
Эритромицин, кларитромицин (но не азитромицин) вызывают удлинение интервала Q-T и усиление кардиотоксических проявлений при совместном применении антигистаминных препаратов (терфенадинаx, астемизолаx), усиливают кардиотоксичность цизапридаx (препарата, стимулирующего моторику ЖКТ).
Эритромицин подавляет метаболизм блокаторов медленных кальциевых каналов (никардипинx и др.), противоопухолевых препаратов (тамоксифен, винбластин), гиполипидемических препаратов группы ингибиторов ГМГ-КоА-редуктазы (ловастатин, симвастатин), снижает метаболизм наркотических анальгетиков (алфентанилаx), ингибиторов протеаз (индинавир), что требует тщательного лекарственного мониторинга и коррекции доз.
Кларитромицин увеличивает AUC для карбамазепина.
Рифампицин, индуцируя ферменты, ослабляет действие антиаритмических препаратов (дизопирамидx, хинидинx), противогрибковых препаратов (кетоконазол, значительно меньше - флуконазола), снижает действие производных бензодиазепина (мидазолам), блокаторов медленных кальциевых каналов (особенно верапамила, дилтиазема, нифедипина, пероральных контрацептивов, глюкокортикоидов, иммунодепрессантов (циклоспорин), уменьшает концентрации макролидов в сыворотке крови (кларитромицин), ингибиторов протеаз (индинавир и др.), анальгетиков (гидрокодонx, фентанил), антиаритмических средств (мексилетинx), β-адреноблокаторов (пиндолол), теофиллина, фенитоина, непрямых антикоагулянтов (варфарин).
Препараты группы фторхинолонов ингибируют цитохром P450 1A2, осуществляющий деалкилирование теофиллина.
Оценивая риск взаимодействия препаратов на уровне метаболизма, необходимо оценивать и время (интервал от начала совместного применения), через которое изменяется кинетика ЛС. Так, эритромицин снижает общий клиренс теофиллина через 5 дней совместного применения на 25%, что может привести к появлению НЛР, свойственных передозировке теофиллина; рифампицин через несколько дней может повысить общий клиренс теофиллина на 50-75%, что способно увеличить симптомы бронхиальной обструкции.
Взаимодействие между антибиотиками
Комбинацию АМП выбирают с целью расширения спектра антибактериальной активности и (или) для усиления терапевтического эффекта. Синергидный эффект возникает при сочетании двух препаратов бактерицидного действия, двух бактериостатических препаратов, бактерицидного препарата (нарушающего функцию цитоплазматической мембраны) с бактериостатическим. Антагонистический эффект наблюдается при назначении бактериолитических препаратов, нарушающих синтез стенки микроорганизма (т.е. действующих только в фазу деления клетки), с бактериостатическими препаратами (блокирующими фазу деления).
Помимо фармакодинамического взаимодействия химиопрепаратов следует учитывать возможность их физико-химической (фармацевтической) несовместимости, что, однако, не является противопоказанием к их комбинированному назначению (например, пенициллины и гентамицин вводят в разных шприцах и капельницах). Несовместимы в одних растворах следующие препараты:
Лучшее правило - всегда вводить антибиотики отдельно! Комбинированное назначение антибиотиков применяют:
-
для повышения антибактериальной эффективности (при лечении тяжелых инфекций до установления бактериологического диагноза);
-
замедления развития резистентности при хронической инфекции (туберкулез, лепра, хронический бронхит);
-
снижения дозы АМП с выраженным токсическим действием без уменьшения эффективности лечения.
Примеры рациональных комбинаций АМП приведены в табл. 26.28.
Таблица 26.28. Фармакодинамическое взаимодействие антибиотиков
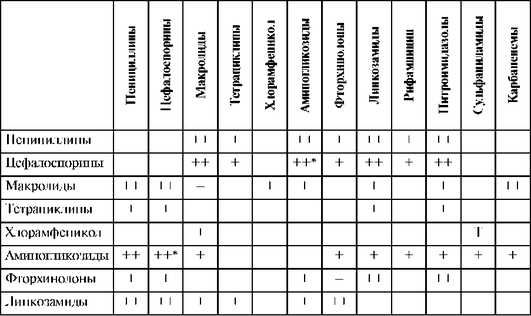
Окончание табл. 26.28
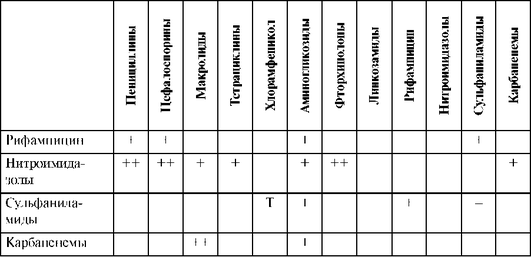
Примечания: ++ - Наиболее рациональные и часто используемые комбинации с хорошо документированной эффективностью.
+ - Комбинация рациональна при ряде показаний, но имеет меньший опыт применения.
Т - Опасное увеличение токсичности.
* Возможно потенциирование нефротоксичности.
Выведение. Взаимодействие лекарственных средств на уровне выведения
Основные пути элиминации ЛС - экскреция и метаболизм. ЛС выводятся из организма преимущественно почками. Другие органы, ткани и жидкости организма играют в этом процессе меньшую роль: легкие, грудное молоко (ко-тримоксазол, пенициллины, метронидазол, тетрациклины), пот, слезы и выделения половых органов (пациент поднимает тревогу, если не был заранее предупрежден, что рифампицин окрашивает мочу, слезную жидкость и другие жидкости организма в оранжево-красноватый цвет). Некоторые антибиотики выводятся в высокой концентрации с желчью: рифампицин, тетрациклины, эритромицин, фторхинолоны и другие, часть из них затем рециркулирует (после повторного всасывания выводятся с желчью, поддерживая высокую концентрацию ЛС в кишечнике). Эта фармакокинетическая характеристика антимикробного ЛС позволяет выбирать антибиотик для лечения холецистита и холангита, кишечных инфекций. Некоторые ЛС после выведения с желчью рециркулируют и окончательно в высокой концентрации выводятся почками (тетрациклины), другие инактивируются в кишечнике (при рециркуляции неактивный метаболит хлорамфеникола реактивируется посредством гидролиза в кишечнике).
Выделение препарата почками происходит главным образом благодаря трем процессам:
Примером препаратов, клиренс которых соответствует СКФ, служит (после поправки на связывание с белками) гентамицин. Поскольку креатинин очищается в основном фильтрацией, измерение скорости почечного клиренса важно для оценки скорости клиренса этих ЛС (проба Реберга). На клиренс также могут влиять два других механизма: секреция и реабсорбция, но в этом случае их действия уравновешивают друг друга. При беременности СКФ увеличивается на 70% и препараты, которые выделяются преимущественно почками, элиминируются быстрее (во время беременности увеличивается клиренс ампициллина).
Некоторые препараты препятствуют активной канальцевой секреции, что служит основой взаимодействия некоторых из них. Так, пробенецидx препятствует активной секреции пенициллинов, пролонгируя их действие; фуросемид блокирует секрецию аминогликозидов, что при длительном применении указанной комбинации может привести к появлению органотоксических эффектов - ототоксическому и нефротоксическому, вплоть до развития нефронекроза; фуросемид подавляет также клиренс ампициллина и цефалоспоринов.
Фармакокинетические характеристики антибиотика (процент выведения ЛС почками в неизмененном виде или в форме активных метаболитов) позволяют выбрать препарат для лечения инфекций мочевыводящих путей (большая часть современных антибиотиков). При низкой доле выведения почками и высокой доле выведения с желчью антибиотик не может быть применен для лечения инфекций почек (препараты группы эритромицина).
При выборе препарата для лечения инфекций мочеполовых органов следует учитывать кислотность мочи. В зависимости от влияния кислотности мочи на активность различают следующие антибиотики.
-
АМП, эффективные при кислой реакции мочи (рН 5,0-6,5): пенициллины, тетрациклины, новобиоцинx, 8-оксихинолины, хинолины, рифампицин, нитрофурантоин (Фурадонин*), фуралтадонx.
-
АМП, эффективные при щелочной реакции мочи (рН 7,5-8,5): макролиды, линкомицин, аминогликозиды.
-
АМП, эффективность которых не зависит от рН мочи: хлорамфеникол, полимиксины, цефалоспорины, ристомицинx, ванкомицин, нитрофурал (Фурацилин*), фуразолидон, циклосерин.
Для подкисления мочи используют аскорбиновую кислоту, кальция хлорид, для подщелачивания - содовое питье, щелочную минеральную воду.
В последние годы существенно расширилась информация о необходимости изменения режима дозирования антибиотиков при почечной недостаточности (снижение дозы препарата или удлинение интервала между введением ЛС) в зависимости от уровня снижения клиренса креатинина или уровня креатина в сыворотке крови, при проведении гемодиализа, а по вновь создаваемым антибиотикам резерва - при пересадке почки.
Следует учитывать, что даже при нормальном клиренсе креатинина и концентрации мочевины в крови у пожилых людей почечный резерв снижен, и после 70 лет необходимо уменьшение доз препарата.
Скорость почечной элиминации препаратов снижается при дегидратации, ХСН, артериальной гипотензии, задержке мочи.
Выделяют антибиотики, режим введения которых корригируют:
Взаимосвязь фармакокинетики и фармакодинамики
Связь терапевтического эффекта с концентрацией ЛС в сыворотке крови - важная проблема современной медицины, позволяющая обосновать наиболее эффективный режим дозирования и применения лекарственного препарата. Успехи клинической фармакологии последних десятилетий позволили вплотную подойти к количественному анализу закономерностей, связывающих терапевтические или токсические эффекты с концентрацией препарата в крови и тканях.
Для многих препаратов довольно точно установлена связь концентраций ЛС в крови с выраженностью фармакодинамических эффектов (антиаритмические средства, сердечные гликозиды, психотропные средства и др.), и на ней основан фармакокинетический контроль при фармакотерапии различных заболеваний.
Труднее установить количественную зависимость между концентрациями ЛС в крови или тканях и выраженностью терапевтического эффекта в клинической химиотерапии.
Микроорганизмы (бактерии) представляют собой достаточно динамичную систему. Степень воздействия антибактериального средства на микроорганизмы количественно описывается МПК, а также минимальной бактерицидной концентрацией, которые сами по себе зависят от вида микроорганизма и его резистентности.
Кроме того, значения МПК определяют на основании данных, полученных in vitro, в то время как инфекционный процесс протекает в организме человека под влиянием многих факторов, изменяющих величину фармакодинамического эффекта (локализация инфекции, рН в месте инфекции, величина микробной нагрузки, состояние защитных сил макроорганизма, синергизм или антагонизм антибактериальных средств).
Ранние химиотерапевтические исследования связи фармакокинетики и фармакодинамики антибиотика сводились к установлению зависимости между концентрацией препарата в крови и выраженностью его токсических эффектов. В последние годы на основании экспериментальных исследований in vitro и in vivo была показана возможность установления количественной зависимости между концентрациями антибактериальных средств в крови или тканях и выраженностью клинического эффекта. Эта зависимость имеет различные характеристики для разных классов противомикробных средств.
Эффективность антибиотиков во многом определяется фармакокинетическими параметрами этих ЛС, такими как отношение ПК препарата к величине МПК (ПК/МПК), отношение AUC к величине МПК (AUC/МПК) и время, в течение которого концентрация препарата превышает величину МПК (T > МПК). С другой стороны, эффективность препарата может в большей степени зависеть от дозы (дозозависимый эффект) или от продолжительности действия препарата, кроме того, ряд антибиотиков обладает пролонгированным эффектом - так называемым постантибиотическим действием.
По противомикробной активности все антибактериальные средства можно разделить на три основные группы (табл. 26.29).
-
Препараты, эффективность которых зависит от их концентрации и постантибиотического эффекта. Эффективность этих ЛС в основном определяется величиной соотношений AUC/МПК илиПК/МПК.
-
Препараты, эффективность которых зависит от продолжительности действия, и которые имеют минимальное постантибиотическое действие. В этом случае основной параметр эффективности - отношение T > МПК.
-
Препараты, эффективность которых определяется как продолжительностью действия, так и концентрацией. Основной фармакокинетический / фармакодинамический параметр эффективности ЛС этой группы - соотношение AUC/МПК.
Таблица 26.29. Параметры фармакокинетики / фармакодинамики, определяющие эффективность различных антибиотиков in vivo
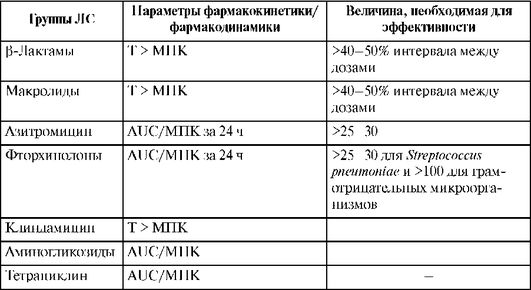
Фармакокинетика и фармакодинамика антибактериальных лекарственных средств и оценка их эффективности
Для β-лактамных антибиотиков характерно бактерицидное действие, которое проявляется в концентрациях, превышающих МПК. При дальнейшем увеличении максимальных концентраций антибиотика скорость гибели бактерий не изменяется. Общее количество убитых микроорганизмов находится в зависимости от времени, в течение которого концентрация антибиотика превышает значение МПК.
Учитывая отсутствие у β-лактамных антибиотиков клинически значимого постантибиотического эффекта, для достижения эффекта наиболее важно поддержание между введениями антибиотика концентраций в сыворотке крови, превышающих МПК. На основании экспериментальных исследований было установлено, что для достижения адекватного клинического эффекта концентрации, превышающие МПК, должны поддерживаться в течение как минимум половины интервала между дозами при применении β-лактамных антибиотиков, т.е. (T > МПК) = 40-50%.
После того как концентрация препарата превысит величину МПК возбудителя, дальнейшее увеличение концентрации теряет смысл, так как эффективность этих препаратов зависит в основном от показателя T > МПК. В тех случаях, когда T > МПК более 40-50% от интервала между очередными дозами, применение β-лактамов эффективно более чем в 80% случаев.
Практически у всех β-лактамных антибиотиков величина T > МПК90 по отношению к пневмококку составляет >40% от интервала дозирования. При инфекциях, вызванных устойчивыми штаммами Streptococcus pneumoniae, только цефтриаксон сохраняет эффективную величину T > МПК90. В отношении Haemophilus influenzae эффективные концентрации в крови поддерживаются при применении цефиксима и цефтриаксона (табл. 26.30).
Таблица 26.30. Величина времени, превышающего МПК90 (T > МПК) для некоторых пероральных β-лактамных антибиотиков
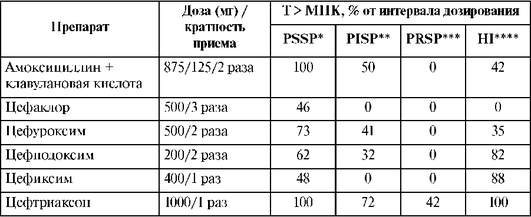
* PSSP - Streptococcus pneumoniae, чувствительные к пенициллинам.
** PISP - S. pneumoniae с промежуточной чувствительностью к пенициллинам.
*** PRSP - S. pneumoniae, резистентный к пенициллинам.
**** HI - Haemophilus influenzae.
Бактерицидное действие карбапенемов, как и других β-лактамов, зависит также не от C (как, например, для аминогликозидов), а от времени поддержания уровня в крови выше МПК для данного возбудителя. Не следует стремиться к тому, чтобы C в крови превышала МПК в 10-15 раз, достаточно ее поддерживать на уровне 2-4-кратных значений. Повышение концентрации β-лактамов выше этого уровня не приводит к увеличению эффекта, т.е. более важна не величина разовой дозы, а кратность введения карбапенемов.
Для аминогликозидов характерно быстрое бактерицидное действие, причем выраженность его прямо зависит от Cmax.
Учитывая наличие у аминогликозидов выраженного постантибиотического действия, для достижения максимального клинического эффекта целесообразно увеличение разовой дозы препарата, при этом интервалы между дозами имеют меньшее значение. Это объясняет современные рекомендации по введению суточной дозы аминогликозидов в один прием. Концентрация аминогликозидов в тканях и плазме крови примерно одинакова.
Фторхинолоны характеризуются быстрым бактерицидным действием, напрямую зависящим от концентрации препарата в крови. В отличие от аминогликозидов, для фторхинолонов характерен умеренный постантибиотический эффект. В связи с этим клиническая эффективность фторхинолонов является функцией не только величины Cmax, но и времени, в течение которого концентрация препарата в крови превышает МПК. В ходе экспериментальных и клинических исследований было установлено, что эффективность фторхинолонов наиболее точно прогнозируется интегральным параметром, представляющим собой отношение AUC к МПК.
В ходе исследований было показано, что адекватный клинический эффект (80% и выше) при применении ципрофлоксацина у тяжелых больных в отделениях интенсивной терапии может быть достигнут при значении AUC/МПК, превышающем 125. Были рассчитаны дозы ципрофлоксацина, при которых ожидается адекватный клинический и бактериологический эффект при выделении различных микроорганизмов. В отношении высокочувствительных штаммов, для которых МПК <0,2 мкг/мл, все изученные дозы ципрофлоксацина (600-1200 мг/сут) могут быть эффективны (AUC/МПК >125).
При выделении менее чувствительных штаммов (Staphylococcus aureus, Pseudomonas aeruginosa) МПК = 0,5 мг/л и для достижения адекватного эффекта (AUC/МПК >125) требуется увеличение суточной дозы ципрофлоксацина до 1,2 г. Доказано, что предупредить возникновение устойчивых штаммов бактерий в процессе лечения можно, если значения AUC/МПК ципрофлоксацина будут превышать 100. Эффективность препаратов этой группы определяется как продолжительностью действия, так и концентрацией. Таким образом, основной фармакокинетический параметр эффективности для этой группы - соотношение AUC/МПК. Эффективное значение AUC/МПК для Streptococcus pneumoniae >25-30, для Haemophilus influenzae >25, а для грамотрицательных бацилл >100. В одном из исследований клиническая и микробиологическая эффективность ципрофлоксацина у 64 пациентов с тяжелой респираторной инфекцией наблюдалась в тех случаях, когда соотношение AUC/МПК было >125, а у 134 пациентов, получавших лечение левофлоксацином, вероятность клинического и микробиологического выздоровления была существенно выше, когда отношение AUC/МПК составляло 100. В отличие от ципрофлоксацина, левофлоксацин демонстрирует высокую эффективность при бактериемии.
Все фторхинолоны достигают эффективного уровня AUC/МПК по отношению к Haemophilus influenzae. Эффективное значение AUC/ МПК для Streptococcus pneumoniae достигается только у новых фторхинолонов с повышенной антипневмокковой активностью (табл. 26.31).
Таблица 26.31. Отношение AUC/МПК (24 ч) для фторхинолонов при лечении пневмококковой инфекции
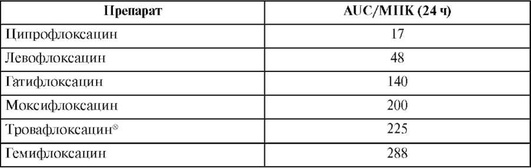
Оценка эффективности, безопасности и продолжительности антибактериальной терапии
Оценка эффективности выбранной терапии, как правило, складывается из оценки клинической и микробиологической эффективности, которую можно проводить как во время лечения (3-5 сут), так и после его завершения.
Клинические критерии
Самое распространенное положение можно представить так: если антибиотик назначен правильно, положительная динамика, хотя бы по отдельным показателям, отмечается уже через 2-3 сут. Через 7-10 дней этиотропное лечение инфекционно-воспалительного процесса обычно можно завершить. Если через 2-3 сут положительная динамика не наблюдается, то необходима коррекция лечения на основании пробелов в противомикробном спектре ранее назначенного препарата и с учетом бактериологических данных.
Эффективность вновь назначенного антибиотика или комбинации антибиотиков оценивают повторно через 48-72 ч, и при положительном клиническом эффекте лечение продолжают еще 7-10 дней; при адекватном выборе средств потребность в третьей смене антибиотиков бывает редко.
Выделяют следующие клинико-лабораторные критерии достаточности антибактериальной терапии, при которых препарат может быть отменен:
-
стабильная гемодинамика (отсутствие артериальной гипотензии, тахикардии);
-
положительная динамика основных симптомов заболевания (кашель, количество мокроты, хрипы в легких, болезненность живота, отделяемое по дренажу и т.д.);
-
отсутствие гнойной мокроты (при бронхолегочных инфекциях), пиурии и лейкоцитурии при мочевых инфекциях (осмотр и обследование инфицированных участков, состояние гнойной раны и т.д.);
Изменение других лабораторных показателей [скорость оседания эритроцитов (СОЭ), фибриноген, С-реактивный белок и др.] не может служить критерием достаточности антибактериальной терапии. Следует подчеркнуть, что к моменту прекращения антибактериальной терапии могут сохраняться некоторые симптомы заболевания (например, кашель, отделение мокроты, хрипы в легких, инфильтрация на рентгенограммах и др.), что не всегда служит поводом для продления применения антибиотика.
Отмена АМП не означает обязательного прекращения лечения больного. При необходимости его можно продолжать с использованием физиотерапевтических методов, иммунотерапии и т.д. Это положение не касается таких состояний, как стафилококковые инфекции (особенно в случае стафилококков, резистентных к метициллину), атипичные пневмонии, вызванные хламидиями, микоплазмой или легионеллой, менингит, эндокардит, фебрильная нейтропения, при которых, вследствие особых условий возникновения и течения инфекций, требуются более длительные курсы антибактериальной терапии даже в случаях быстрого достижения улучшения клинического состояния больного.
При наличии у больного тяжелых сопутствующих заболеваний или снижения иммунитета продолжительность антибиотикотерапии также увеличивают индивидуально.
Бактериологические и иммунобиологические критерии
Динамика бактериологических и иммунологических показателей - посевы патологических материалов с оценкой степени эрадикации (удаления) возбудителя представлены в табл. 26.32.
Таблица 26.32. Микробиологические критерии эффективности проводимого лечения
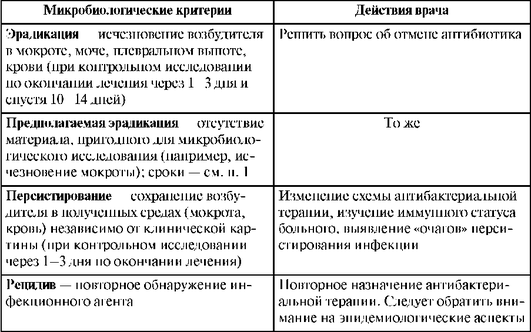
Оценка безопасности антимикробных лекарственных средств
Кроме клинической эффективности проводимого лечения необходимо помнить об обеспечении приемлемого соотношения риск/ эффективность и, следовательно, о проведении оценки безопасности осуществляемой терапии.
Для оценки безопасности применения АМП существуют клинические и лабораторные методы обнаружения НЛР, т.е. любых неблагоприятных событий, возникших в период приема препарата, независимо от того, связаны они с его приемом или нет.
Планируя проведение клинического контроля за выявлением НЛР, врач может использовать любой метод (произвольное сообщение больного, вопрос врача о клинических событиях, вопрос о конкретном ЛС, стандартный опросник). От выбранного метода зависит частота обнаружения НЛР.
Кроме клинического контроля необходимо проводить лабораторный и инструментальный контроль развития НЛР (аудиограмма при применении препаратов с риском развития ототоксического действия). При этом выбор методов контроля зависит от имеющихся сведений о потенциальных НЛР, вызываемых препаратом, и от опыта работы с ним. Например, определение нефротоксического действия аминогликозидов по повышению уровня креатинина более чем на 50% от первоначальных данных на фоне применения антибиотика или установление гепатотоксического действия пефлоксацина при увеличении активности ферментов печени (аланинаминотрансферазы, аспартатаминотрансферазы) более чем в 2,5-3 раза. В зависимости от вида и тяжести НЛР решается дальнейшая тактика лечения - отменить препарат или продолжить его применение при проведении тщательного контроля безопасности.
Нежелательные лекарственные реакции антибактериальных препаратов
Выбор антибиотика необходимо осуществлять с учетом риска развития НЛР. При наличии патологии органа или системы органов антибиотик с соответствующей направленностью НЛР (особенно органотоксических эффектов) выбирать не следует. При отсутствии возможности отказаться от применения антибиотика, обусловливающего высокий риск развития НЛР, необходимо обеспечить безопасный режим дозирования (например, снизить дозу при почечной недостаточности) и контроль за всеми прогнозируемыми НЛР (программа динамического контроля безопасности).
НЛР могут быть связаны с гиперчувствительностью (аллергические реакции, интерстициальный нефрит, васкулит, лихорадка), токсическим действием (нефротоксичность, гепатотоксичность, ототоксичность, в том числе эмбриотоксическое действие, нейротоксичность), желудочно-кишечные реакции, геморрагический синдром, флебиты, электролитные нарушения, нарушение толерантности к алкоголю, а также действие, связанное с подавлением сапрофитной микрофлоры (дисбактериоз, суперинфекция), угнетение иммунитета, развитие вторичных инфекций, гипо- и авитаминозов, реакцией массивного бактериолиза.
Аллергические реакции
Аллергические реакции (немедленные - до 30 мин, быстрые - 1-48 ч, отсроченные - более 48 ч) могут развиваться при применении любых АМП, но наиболее часто наблюдаются на фоне применения β-лактамных антибиотиков и сульфаниламидов. Их возникновение не связано с фармакологическим эффектом и не зависит от дозы препарата (хотя проявления аллергии может усиливаться при повышении дозы). Для них характерно обязательное проявление после повторного назначения того же (или близкого по химической структуре) противомикробного средства (внутригрупповая перекрестная аллергическая чувствительность), при этом латентный период становится короче, а симптомы - более выраженными.
По частоте вызываемых аллергических реакций первое место занимают пенициллины, затем следуют стрептомицин, ванкомицин, амфотерицин В, сульфаниламиды. Гораздо реже аллергические проявления возникают при применении тетрациклинов, цефалоспоринов, нитрофуранов, оксихинолинов, фторхинолонов, рифампицина. В отличие от β-лактамов, аллергические реакции на макролиды встречаются крайне редко.
Токсические реакции
Токсические реакции возникают чаще аллергических; их клинические проявления находятся в прямой зависимости от дозы, пути введения, длительности лечения, особенностей взаимодействия с другими ЛС.
Нейротоксические реакции
Поражение слухового нерва наиболее характерно для аминогликозидов. По уменьшению частоты развития данного осложнения препараты можно расположить в следующем порядке: мономицин®, канамицин, неомицин, стрептомицин. Наименее выражен побочный эффект у нетилмицина. Ототоксическое действие может проявиться даже при местном применении аминогликозидов (особенно у больных с нарушением функций почек). Применение стрептомицина во время беременности может привести к врожденной глухоте. Нарушение слуха может прогрессировать и после отмены препарата. Противопоказано сочетание нескольких аминогликозидов, а также аминогликозидов с другими препаратами, вызывающими нарушение слуха, например салицилатами, хининомx, этакриновой кислотой, фуросемидом. Нельзя повторно назначать аминогликозиды ранее 10-12 дней после окончания курса применения препарата этой группы. Во время лечения необходимо проведение аудиограммы. Лучше не назначать аминогликозиды больным старше 60 лет.
Поражение вестибулярного аппарата вызывают стрептомицин, канамицин, неомицин, гентамицин.
Снижение слуха при применении макролидов возникает очень редко, причем чаще всего это происходит у пациентов с нарушением функций почек, получавших эритромицин и у ВИЧ-инфицированных больных с диссеминированной инфекцией Mycobacterium avium, получавших комбинированную терапию азитромицином и этамбутолом. При лечении макролидами у 0,2-1,3% больных встречаются неврологические расстройства в виде головной боли, головокружения, бессонницы.
Поражение зрительного нерва возможно при приеме стрептомицина, хлорамфеникола, циклосерина, полимиксина В, налидиксовой кислоты. Нарушение зрения также могут вызывать изониазид, этамбутол, этионамид.
Полиневрит, эпилептиформные припадки могут быть вызваны стрептомицином, метронидазолом, полимиксином, амфотерицином В, циклосерином, хлорамфениколом, сульфаниламидом, нитрофураном, оксихинолином, налидиксовой кислотой, фторхинолоном, канамицином, гризеофульвином, хлорамфениколом, изониазидом, этамбутолом.
Головокружение, головная боль, нарушение сна могут быть вызваны фторхинолонами.
Парестезии, головная боль, головокружение, атаксия возможны при приеме полимиксина В, стрептомицина, циклосерина, амфотерицина В.
Особенно часто прямое нейротоксическое действие (галлюцинации, эпилептиформные припадки, судороги и мышечный гипертонус) возникает при интралюмбальном введении бензилпенициллина, стрептомицина, хлорамфеникола.
Развитие нервно-мышечной блокады, паралич дыхания, коллапс могут быть обусловлены полимиксином В (часто), аминогликозидами, линкомицином (при быстром введении больше доз выше 200 мг/кг внутривенно), клиндамицином. Снижение АД, головокружение, тахикардия могут возникнуть при внутривенном введении фторхинолонов.
Нарушение вкуса возможно при применении ампициллина, метронидазола, тетрациклина, судороги - при применении азтреонама, имипенема + циластатина, метронидазола, хинолонов, а также больших доз бензилпенициллина.
Нефротоксические реакции
Нефротоксичность (снижение функций почек с увеличением концентрации в крови мочевины, креатинина) наиболее часто развивается на фоне применения аминогликозидов, ванкомицина, а также может встречаться при использовании полипептидных антибиотиков (полимиксин В, бацитрацин), аминогликозидов, цефалоспоринов - цефалотина, цефалексина (цефамандол может вызывать глюкозурию), амфотерицина В, рифампицина, гризеофульвина, сульфаниламидов, редко - нистатина, налидиксовой кислоты, ампициллина, цефалоспоринов III-IV поколения, тетрациклинов и др. К предрасполагающим факторам относятся: пожилой возраст, артериальная гипотензия, гиповолемия, заболевание печени, предшествующее лечение аминогликозидами, сочетание некоторых ЛС (например, аминогликозидов, цефалоспоринов, ампициллина с петлевыми диуретиками - фуросемидом, этакриновой кислотой).
Риск развития НЛР увеличивается при нарушении выделительной функции почек. Как правило, поражения почек обратимы в ближайшие 2-3 дня после отмены препаратов.
Интерстициальный нефрит (симптомы: гематурия, протеинурия, лихорадка, сыпь, эозинофилия в крови и моче, в 50% случаев - нарушение функций почек) чаще вызывается полусинтетическими пенициллинами (оксациллин).
Гепатотоксические реакции
Гепатотоксические эффекты могут проявляться в двух формах: холестаза и гепатита. Они развиваются на фоне применения противотуберкулезных средств, оксациллина, азтреонама, тетрациклинов, линкозамидов, сульфаниламидов.
Многие препараты, накапливающиеся в желчи в больших концентрациях, могут вызвать поражения печени: гепатомегалию, гипербилирубинемию (тетрациклины, сульфаниламиды), гепатиты (амфотерицин В). Реже эти осложнения могут быть обусловлены приемом полимиксина В, налидиксовой кислоты, линкомицина, нитрофуранов, хлорамфеникола, рифампицина, фторхинолонов, даже пенициллинов и эритромицина.
Макролиды в редких случаях вызывают холестатический гепатит, который нередко протекает под маской острого холецистита. Чаще всего он развивается при лечении эритромицином. Наряду с клиническими проявлениями гепатита, у 2/3 пациентов отмечают эозинофилию, гипербилирубинемию и увеличение активности щелочной фосфатазы.
На фоне лечения карбенициллиномx и тикарциллином у 5-20% пациентов также могут наблюдаться увеличение активности печеночных трансаминаз в крови и гипокалиемия. Рифампицин повышает содержание билирубина и активность аминотрансфераз в крови, что можно устранить без отмены препарата назначением метионина, пиридоксина. Жировая дистрофия печени иногда возникает на фоне высоких доз тетрациклинов.
Даже при нормальном функционировании печени лечение препаратами, оказывающими гепатотоксическое действие, следует проводить на протяжении не более 7-10 дней. В большинстве случаев описанные изменения исчезают после отмены препаратов. Лицам старше 70 лет дозы этих препаратов снижают в 2-3 раза.
Токсическое действие на желудочно-кишечный тракт
Токсическое действие на ЖКТ наблюдается с различной частотой при применении практически всех антибактериальных препаратов, главным образом при приеме внутрь. Токсический гастрит (тошнота, рвота, анорексия, боль в животе, диарея, запор) наиболее часто возникает при приеме тетрациклинов, ампициллина, макролидов, гризеофульвина, амфотерицина В, канамицина, фузидовой кислоты. Обычно эти НЛР не выражены, необходимости в отмене препарата не возникает. Реже встречаются стоматит, эзофагит (тетрациклины), «металлический» привкус во рту (метронидазол).
Псевдомембранозный колит (стафилококковый энтероколит, холероподобный синдром) стал регистрироваться на фоне применения почти всех антибиотиков широкого спектра действия, чаще развивается при назначении линкозамидов (линкомицина, клиндамицина), тетрациклинов, реже - при назначении аминогликозидов, хлорамфеникола, цефалоспоринов. Возникает на фоне антибактериальной терапии вследствие эндогенной суперинфекции (этиологический фактор Clostridium defficile) и сопровождается выраженной диареей (4- 6 раз в сутки и более), кровянистым стулом, схваткообразными болями в животе, лихорадкой (39-40 °C), лейкоцитозом, в тяжелых случаях - артериальной гипотензией, альбуминурией, профузной диареей с выделением пленчатого материала. Возможно образование язв на слизистой оболочке кишечника.
При появлении этих симптомов в легких случаях достаточно отмены препарата и назначение ионообменных смол (колестираминx, колестиполx), а в тяжелых случаях показано возмещение потери жидкости, электролитов и белков, назначение антибиотиков, активных против Clostridium defficile (ванкомицин в дозе 125-500 мг 4 раза в сутки, или бацитрацин в дозе 25 000 ЕД 4 раза в сутки в течение 7-10 дней, или метронидазол по 500 мг 3 раза в сутки), сорбентов и эубиотиков. Нельзя применять препараты, тормозящие перистальтику кишечника, в том числе лоперамид (Имодиум*).
Принимаемые внутрь АМП снижают активность панкреатических ферментов, что может вызвать диарею.
Суббактериостатические концентрации практически всех антибиотиков уменьшают антигенную и иммунизирующую активность микроорганизмов.
Гентамицин снижает количество иммунокомпетентных клеток в селезенке и титр агглютининов в сыворотке крови (иммунодепрессивный эффект), поэтому при его применении, особенно для лечения сепсиса, целесообразно назначить иммунокорректоры. Хлорамфеникол подавляет образование антител.
Геморрагический синдром
Геморрагический синдром характерен для цефалоспоринов II- III поколения, имеющих в основе N-метилтиотетразольное кольцо (цефамандол, цефотетанx, цефоперазон нарушают всасывание витамина K в кишечнике). Антисинегнойные пенициллины (карбенициллинx, тикарциллин, уреидопенициллины) нарушают функцию мембран тромбоцитов. Бензилпенициллин, карбенициллинx, тикарциллин в 1-2% случаев могут вызывать кровоточивость за счет снижения агрегации тромбоцитов (подобно ацетилсалициловой кислоте). Кожные реакции и лихорадка (аллергического генеза) встречаются в 2-4% случаев применения пенициллинов, наиболее часто - на фоне ампициллина (4-18%).
Нейтропения / агранулоцитоз в единичных случаях возникает при применении антисинегнойных пенициллинов, нитрофуранов, сульфаниламидов, рифампицина, чаще - при применении хлорамфеникола.
Апластическая анемия наиболее часто наблюдается при применении хлорамфеникола и характеризуется нарушением функции костного мозга с развитием лейкопении, тромбоцитопении, анемии.
Гемолиз эритроцитов может развиться под действием различных АМП: β-лактамных антибиотиков, ко-тримоксазола и триметоприма (аутоиммунный гемолиз), а на фоне наследственного дефицита в эритроцитах Г-6-ФД - при применении сульфаниламидов, нитрофуранов, фторхинолонов, ко-тримоксазола и рифампицина.
Флебиты могут возникнуть при внутривенном применении практически всех противомикробных средств, но чаще (в порядке убывания) при инфузиях монобактамов, тетрациклинов, ванкомицина, полимиксина В, цефалоспоринов. При увеличении разовой дозы эритромицина (до 1 г) или быстром введении (менее 30 мин) усиливается раздражающее действие антибиотика на стенку сосуда и повышается риск возникновения флебита.
Известны случаи токсического влияния хлорамфеникола на функцию надпочечников.
Эмбриотоксическое действие антибиотиков
Большие дозы тетрациклина в поздние сроки беременности могут вызвать острую желтую дистрофию печени (особенно при парентеральном введении). Даже небольшие дозы тетрациклинов (концентрация в пупочных сосудах около 50-60% от уровня в крови матери) в поздний период беременности могут вызвать окрашивание зубов плода, их гипоплазию, а также замедленное развитие костного скелета (тетрациклины запрещены к применению у беременных).
Пенициллины (особенно полусинтетические) и цефалоспорины также проникают через плаценту, создавая в тканях плода терапевтическую концентрацию (токсических действий при этом обычно не наблюдается, возможна аллергизация плода). Способность полусинтетических пенициллинов проникать через плацентарный барьер находится в обратной зависимости от их способности связываться с белками плазмы крови. При аллергии к пенициллинам, помимо цефалоспоринов, может использоваться эритромицин.
Стрептомицин быстро проходит через плаценту (его концентрация в крови плода около 50%), может приводить к различным поражениям нервной системы (в том числе ототоксическое действие), микромиелии, различным нарушениям в строении скелетных костей. Более безопасны гентамицин и канамицин, но и их можно назначать только по жизненным показаниям (для них также характерно ототоксическое и нефротоксическое действие).
Несмотря на высокую токсичность хлорамфеникола, сообщений об его эмбриотоксическом влиянии нет (концентрация в крови плода составляет около 30-80% от таковой в крови матери). У новорожденных могут отмечаться явления коллапса, «серого синдрома».
Не следует использовать в последнем триместре беременности сульфаниламиды (особенно препараты длительного действия), способные интенсивно связываться с белками, поскольку они, вытесняя билирубин из связи с белками, могут вызвать желтуху новорожденных. Кроме того, они (а также нитрофураны) способствуют развитию гемолитической анемии у детей с дефицитом Г-6-ФД. При беременности также не рекомендуют использовать ко-тримоксазол из-за возможности нарушения обмена фолиевой кислоты в организме матери и ребенка.
Из-за высокого риска эмбриотоксического действия метронидазол и триметоприм не применяют в I триместре беременности.
При беременности можно применять:
-
1-3-й месяц беременности: пенициллины, цефалоспорины, линкомицин, фузидовую кислоту;
-
4-7-й месяц беременности: пенициллины, цефалоспорины, линкомицин, фузидовую кислоту, нитрофураны, налидиксовую кислоту (Невиграмон*);
-
последние недели беременности: пенициллины, цефалоспорины, линкомицин, фузидовую кислоту.
Биологическое действие
Суперинфекция
Суперинфекция проявляется чрезмерным ростом условно-патогенной микрофлоры, устойчивой к данному препарату (на фоне подавления нормальной микрофлоры).
Одна из разновидностей суперинфекции - часто возникающий кандидоз (кожи, слизистых оболочек, внутренних органов). У истощенных больных и больных хроническими заболеваниями кандидоз может приобрести системное течение и вызвать летальный исход. Решение об отмене антибактериальных средств принимают строго индивидуально в зависимости от тяжести основного процесса, выраженности и распространенности кандидоза (обнаружение мицелия при микроскопии нативных препаратов мочи, мокроты, экссудатов, увеличение количества мицелиальных элементов при повторных обследованиях, появление клинических признаков висцеральных поражений).
Гиповитаминозы
Витамины В2, В6, В12, РР, пантотеновая кислота вырабатываются кишечной микрофлорой, которая может быть подавлена при пероральном приеме антибиотиков широкого спектра действия. Гиповитаминозы проявляются ухудшением общего состояния, анорексией, поражением кожи и слизистых оболочек, неврологическими расстройствами.
Реакция массивного бактериолиза
Реакция массивного бактериолиза (реакция Яриша-Герксгеймера) - терапевтический шок. Массивный бактериолиз развивается быстро, обычно в начале лечения при введении антибиотиков в больших дозах. Он проявляется потрясающим ознобом, лихорадкой, тахикардией, проливным потом, диареей. В тяжелых случаях может понизиться температура тела. Возможны развитие коллаптоидной реакции, потеря сознания, олигурия и анурия. Указанную реакцию наблюдают при лечении брюшного тифа, коклюша, сифилиса, бруцеллеза, лептоспироза. Образование эндотоксинов характерно для сальмонелл, шигелл, бруцелл, кишечной палочки, синегнойной палочки, протея, возбудителя коклюша, пастерелл, спирохет, микобактерий.
Нарушение толерантности к этанолу
Нарушение толерантности к этанолу развивается при применении метронидазола, хлорамфеникола и цефалоспоринов II-III поколения, имеющих в своей структуре метилтиотетразольное кольцо (применение цефамандола, цефоперазона, цефотетанаx, цефметазола на фоне потребления алкоголя вызывает тошноту, рвоту, головокружение, головную боль, гипотензию, потливость). Тетурамовый эффект возникает при совместном применении нитрофуранов с этанолом.
Фоточувствительность
Фоточувствительность может наблюдаться при применении фторхинолонов, реже - тетрациклинов, сульфаниламидов. Она проявляется потемнением кожи на открытых участках тела под воздействием солнечных лучей (вплоть до ожогов), развитием крапивницы.
Нежелательные лекарственные реакции комплексной природы
НЛР комплексной природы (аллергические, токсические, дисбактериоз, гиповитаминоз, суперинфекции, нарушение синтеза белка):
-
колоаноректальный синдром: колит, проктит, болезненные дефекации, тенезмы, слизисто-кровянистые испражнения. Часто возникают при лечении тетрациклином, реже - хлорамфениколом (Левомицетин*) и эритромицином. Симптомы появляются в первые 4-5 дней лечения;
-
генитоаноректальный синдром: зуд кожных покровов и слизистых оболочек половых органов, переходящий в баланит или вульвовагинит.
Стоимость лечения и возможности снижения затрат на антибактериальную терапию
Экономические показатели антибактериальной терапии начали интенсивно изучать сравнительно недавно, что обусловлено увеличением количества применяющихся в клинической практике антибиотиков, а также их стоимости. По статистике, в России 25-35% финансовых затрат больниц приходится на АМП (за рубежом - до 50-60%).
Соотношения стоимости и эффективности лечения изучают с помощью фармакоэкономических исследований. Следует различать понятия «стоимость антибиотика», подразумевающего его закупочную цену, и «стоимость антибактериальной терапии». Последнее понятие гораздо шире, оно включает в себя несколько показателей: стоимость самого антибактериального ЛС, стоимость его введения, стоимость дополнительной антибактериальной терапии при ее неэффективности и (или) развитии НЛР, стоимость пребывания больного в стационаре. Таким образом, при учете стоимости антибактериальной терапии необходимо учитывать прямые и косвенные затраты.
Существует ряд подходов к снижению затрат на антибактериальную терапию в стационаре при сохранении высокой клинической эффективности:
Стоимость внутривенного лечения антибиотиками (в частности, макролидами) достаточно высока как за счет более высокой цены парентеральных форм (в 6-10 раз), так и вследствие затрат на расходуемые материалы: шприцы, капельницы, стерильные растворы. Поэтому получила распространение так называемая ступенчатая (step-down) терапия, при которой лечение начинается с внутривенного применения антибиотика, а по достижении клинического эффекта (обычно через 2-3 дня) пациент переводится на пероральную терапию тем же или другим лекарственным средством.
Весьма существенным является вопрос о сроках парентеральной бактериальной терапии и критериях возможного перехода на таблетированные ЛС. К обязательным условиям относятся улучшение клинического состояния больного, нормализация температуры тела и показателей лейкоцитарной формулы. Считается, что убедительным доказательством возможности перехода на прием антибиотика внутрь служит сохранение нормальной температуры тела в течение 8-16 ч. Обычно продолжительность внутривенной антибактериальной терапии составляет 2-3 дня, а последующей пероральной химиотерапии - 5-7 дней.
Ступенчатая терапия возможна только при заведомо хорошей всасываемости, и при правильном ее проведении эффективность сравнима с парентеральным лечением. При этом уменьшается частота НЛР, в первую очередь флебитов. Стоимость ступенчатой терапии значительно ниже цены полного курса парентерального лечения. Ступенчатую монотерапию можно проводить макролидами (эритромицин, кларитромицин, спирамицин), фторхинолонами (офлоксацин и ципрофлоксацин), цефалоспоринами и так далее, т.е. препаратами, которые выпускаются в двух лекарственных формах: для внутривенного введения и для приема внутрь. Лекарственное средство для приема внутрь должно иметь высокую биодоступность. Ступенчатую терапию нельзя применять при менингите, инфекционном эндокардите, полирезистентности возбудителя к антибиотикам, нарушении всасывания в ЖКТ.
Кроме снижения прямых расходов на лечение (стоимость лекарств и расходуемых материалов), ступенчатая терапия снижает затраты труда медицинских сестер на выполнение назначений, улучшает переносимость лечения. За рубежом ступенчатая терапия способствует сокращению сроков пребывания в стационаре. Больных пневмонией выписывают еще до завершения лечения - в течение первых двух суток пероральной терапии, если температура тела и состояние больных остаются стабильным.