Нефрология. Клинические рекомендации / под ред. Е. М. Шилова, А. В. Смирнова, Н. Л. Козловской. - М. : ГЭОТАР-Медиа, 2016. - 816 с. - ISBN 978-5-9704-3714-8. |
Аннотация
Первые национальные клинические рекомендации по нефрологии подготовлены коллективом экспертов, в который вошли не только специалисты-нефрологи ведущих нефрологических школ России, но и представители других медицинских специальностей, тесно сотрудничающие с нефрологами, - кардиологи, эндокринологи, инфекционисты, педиатры, генетики. Издание содержит информацию по наиболее распространенным нефрологическим заболеваниям и синдромам. Представленные в нем клинические рекомендации детально описывают действия врача по диагностике, лечению, профилактике и реабилитации пациентов. Соблюдение международной методологии при подготовке клинических рекомендаций гарантирует их современность, достоверность, обобщение лучшего мирового опыта и знаний, обеспечивает возможность практического применения. Именно поэтому клинические рекомендации обладают преимуществами по сравнению с традиционными источниками информации (учебники, руководства, монографии), что позволит врачу в короткие сроки принимать обоснованные решения в сложных клинических ситуациях. Клинические рекомендации по нефрологии предназначены не только практикующим врачам-нефрологам, но и терапевтам, педиатрам, представителям смежных дисциплин. Они также могут использоваться для обучения студентов старших курсов и клинических ординаторов терапевтических специальностей.
Кодирование по Международной классификации болезней 10-го пересмотра
Класс XIV: Болезни мочеполовой системы.
-
N10-N16 Тубулоинтерстициальные болезни почек.
-
N14.1 Нефропатия, вызванная другими лекарственными средствами, медикаментами или биологически активными веществами.
-
N14.2 Нефропатия, вызванная неуточненным лекарственным средством, медикаментом или биологически активным веществом.
-
N16.4 Тубулоинтерстициальное поражение почек при системных болезнях соединительной ткани.
Класс XIV: Болезни костно-мышечной системы и соединительной ткани.
Определение
Хронический тубулоинтерстициальный нефрит (ХТИН) представляет собой хроническое заболевание почек, развивающееся в ответ на длительное воздействие экзо- и/или эндогенных факторов и проявляющееся воспалительными изменениями тубулоинтерстициальной ткани с развитием интерстициального фиброза и тубулярной атрофии с частым развитием хроническая почечная недостаточность (ХПН).
Эпидемиология
Вопрос распространенности ХТИН, так же как и острого тубулоинтерстициального нефрита (ОТИН), является одним из самых сложных. Существенные различия в распространенности нефритов микробного и лекарственного генеза в России и за рубежом определяются несовершенством технологий выявления и регистрации этой патологии, несогласованностью диагностических критериев, а иногда неспецифичностью клинических проявлений некоторых форм интерстициальных нефритов. По данным ряда центров, при проведении пункционной нефробиопсии ХТИН регистрируется в 1,8-2,5% случаев. Однако по данным клинических исследований удельный вес ХТИН выше и колеблется от 4 до 12%.
При анализе причин хронической интерстициальной патологии почек было показано, что в 63,4% случаев нефрит развился вследствие хронического лекарственного воздействия (НПВС, анальгетики, фуросемид и др.), в 14,6% случаев - вследствие бактериального воздействия, в 10,8% - обструктивного воздействия, в том числе мочекаменной болезни, пиелоренального рефлюкса, стриктуры мочеточника, аберрантных сосудов, в 3,2% - вследствие длительного экзогенного токсического воздействия, в 8% - неясного генеза.
Этиология и патогенез
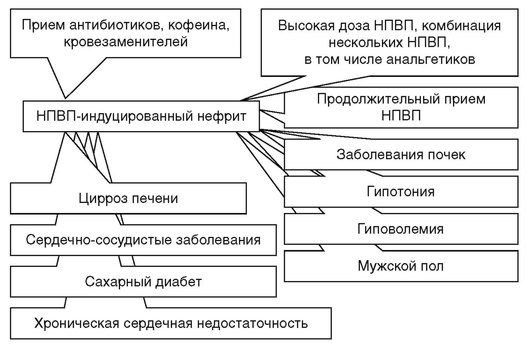
Причинами, приводящими к развитию ХТИН, могут быть инфекционные процессы, вызванные бактериями, вирусами, метаболические нарушения, тяжелые металлы, заболевания с иммунным генезом, неопластические заболевания, радиация, наследственные болезни почек. Крайне редко наблюдаются наследственные формы тубулоинтерстициального нефрита, связанные в частности с мутацией генов муцина-1, уромодулина, семейная хроническая интерстициальная нефропатия с гиперурикемией и др. Анальгетики и НПВС являются наиболее частыми причинами развития лекарственного ХТИН. ХТИН лекарственного генеза страдают преимущественно лица старше 40-45 лет. 65% лиц с анальгетической нефропатией составили лица трудоспособного возраста. Многие исследователи отмечают, что у женщин лекарственная патология почек развивается чаще, что обусловлено более частым применением анальгетиков и НПВС. На рис. 1 представлены факторы риска, способствующие развитию ХТИН наравне с воздействием причинного фактора.
Нередко ХТИН представляет собой тубулоинтерстициальный фиброз на фоне рецидивирующих ОТИН, например, вследствие применения анальгетиков, и морфологические признаки воспаления увидеть в биоптате не представляется возможным, что у некоторых специалистов вызывает дефиниционные споры. У большинства больных при длительном приеме анальгетиков, НПВС или других препаратов как таковых эпизодов ОТИН не наблюдается, постепенно формируется тубулоинтерстициальный фиброз, а в биоптате наблюдаются невыраженные признаки воспаления в виде легкой лимфогистиоцитарной инфильтрации. Монотонное течение без эпизодов ОПП также характерно для поражения почек при сухом синдроме Шегрена.
Помимо НПВС и анальгетиков причинами развития ХТИН могут стать и другие препараты. Наиболее часто это антибиотики. Также описаны случаи ХТИН при терапии варфарином, тиазидными диуретиками, индапамидом, месалазином, ранитидином, циметидином (табл. 1).
| Класс лекарств | Примеры |
|---|---|
Антибиотики |
Аминогликозиды, цефалоспорины, фторхинолоны (ципрофлоксацин), этамбутол, изониазид, макролиды, пенициллины, рифампицин, сульфониламиды, тетрациклин, ванкомицин |
Противовирусные препараты |
Ацикловир, интерфероны |
НПВС, анальгетики |
Практически все представители НПВС, фенацетин, метамизол натрия |
Диуретики |
Фуросемид, тиазидные, индапамид, триамтерен |
Антисекреторные препараты |
Блокаторы водородной помпы (омепразол, лансопразол), Н2-гистаминоблокаторы (ранитидин, циметидин, фамотидин) |
Гипотензивные препараты |
Амлодипин, каптоприл, дилтиазем |
Разное |
Аллопуринол, азатиоприн, карбамазепин, клофибрат, фенитоин, контрасты для ангиографии, препараты на основе поливинилпирролидона, ингибиторы кальцинейрина (циклоспорин) |
Патологическое воздействие на почку реализуется посредством вазоспастических реакций, прямого тубулотоксического действия, активации коллагенообразования (усиливается синтез коллагена 1-го типа), цитокиновой активации с сосудистым ремоделированием, стимулированием эпителиально-мезенхимального перехода и атрофией эпителия канальцев.
Клиническая картина
Клиническая картина ХТИН однотипна и состоит из гипертензионного, мочевого синдромов и синдрома почечной дисфункции. При ХТИН может наблюдаться полиурия, обусловленная нарушением концентрационной способности почек, что можно рассматривать в рамках почечной формы несахарного диабета.
Частота гипертензионного синдрома составляет 33,3-60%, а по некоторым данным, повышение систолического АД наблюдается у 81%, диастолического АД - у 90,5% лиц с гистологически верифицированным ХТИН, развившимся на фоне длительного приема анальгетиков.
ХПН развивается у 40-64% пациентов с лекарственным ХТИН.
Мочевой синдром представлен гематурией и/или невысокой протеинурией. При гистологически подтвержденном ХТИН гематурия выявляется у 81% пациентов, протеинурия - у 90,5%. При постановке диагноза без привлечения нефробиопсии частота протеинурии колеблется от 13% до 40%.
В моче у больных с лекарственным ХТИН нередко наблюдается лейкоцитурия, которая представлена лимфоцитурией и эозинофилурией (60-70% случаев).
Полиурия встречается в 47,6% случаев ХТИН вследствие приема НПВС.
Морфологические изменения, характерные для хронического интерстициального нефрита, как правило, не имеют этиологической метки и в большинстве случаев не способствуют установлению причины их развития. Исключение могут составить интерстициальные изменения при подагре, миеломе, парвовирусной инфекции в случае ее идентификации в ткани биоптата у лиц с иммунодепрессией.
Анализ исследований по проблеме морфологической диагностики интерстициальных нефритов показал, что основными проявлениями являются:
Сосочковый кальциноз является проявлением чаще анальгетической нефропатии и обусловлен капиллярным некрозом или дистрофией сосочковой зоны с последующим фиброзированием и импрегнацией солями кальция. Частота развития колеблется, по разным данным, от 0,2 до 5,8%, редко - до 50%.
Самым ранним поражением при ХТИН является капиллярный склероз, затем возникают центральные некрозы сосочков, позже - тубуло-интерстициальный нефрит с корковой атрофией. При этом морфологические изменения на уровне коры возникают в ответ на обструкцию канальцев в некротизированном мозговом слое.
Диагностика
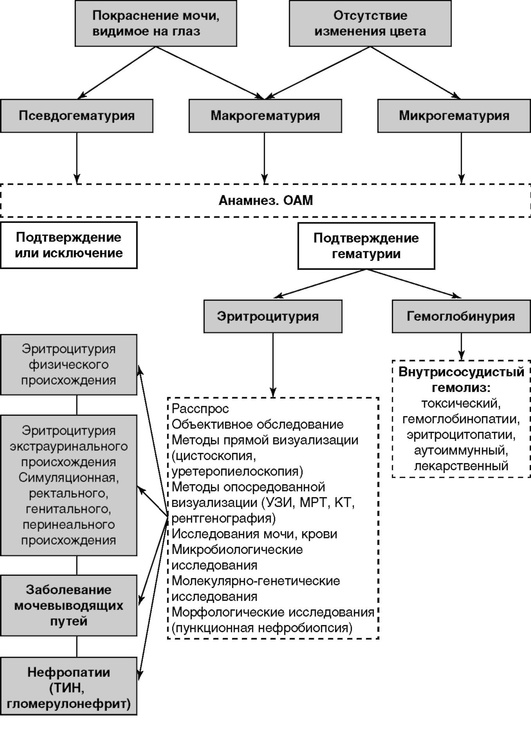
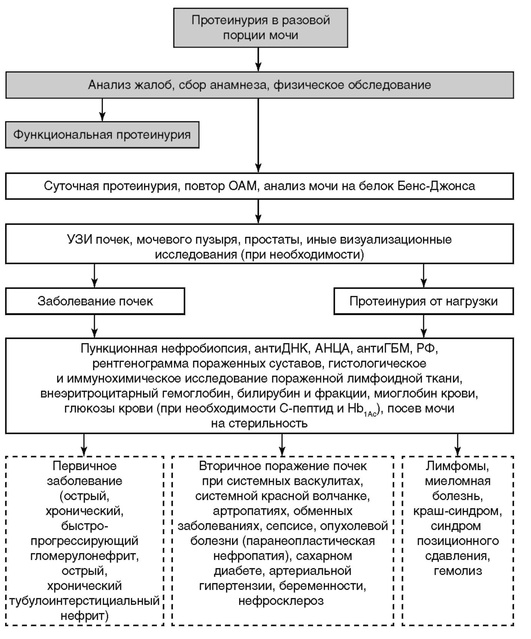
Обязательными методами диагностики являются общий анализ мочи или анализ мочи с помощью тест-полоски и количественные методы оценки мочевого осадка (анализ по Нечиперенко или Аддису-Каковскому или Амбурже), а также оценка уровня креатинина крови с определением СКФ, УЗИ почек. Также показано проведение исследований, направленных на исключение других форм почечной патологии, урологической патологии, проявляющейся гематурией (простатит, опухоль предстательной железы, мочевого пузыря, почки, мочекаменная болезнь, нефроптоз, врожденные аномалии верхних мочевых путей обструктивного типа и др.). Важным методом ранней диагностики ХТИН лекарственного генеза является регулярный мониторинг анализа мочи и СКФ.
Рекомендация 1. Больным с синдромом хронической боли, получающим НПВС и/или анальгетики, необходимо оценивать СКФ и мочевой осадок не реже одного раза в год вне зависимости от кратности и дозы принимаемых препаратов (1В).
Рекомендация 2. При постановке диагноза НПВС/анальгетик-ассоциированного ХТИН следует учитывать основные и дополнительные критерии (табл. 2). При этом обязательно учитывается продолжительность терапии препаратом и рассчитывается примерная суммарная доза приема препарата за весь период его приема (2С).
Основные критерии |
Употребление НПВС (неселективных или селективных), в том числе фенацетин- или метамизолсодержащих анальгетиков на протяжении 12 мес и более в установленной суммарной дозе* Наличие стойкого мочевого синдрома длительностью более 3 мес в виде эритроцитурии или микроальбуминурии или протеинурии не более 3000 мг/сут или β2-микроглобулинурии или абактериальной лейкоцитурии, резистентной к антибактериальной терапии |
Дополнительные критерии |
Снижение СКФ менее 60 мл/мин на протяжении 3 мес и более АГ Гипоизостенурия Признаки интерстициального нефрита и/или тубулоинтерстициального фиброза по данным нефробиопсии Кальцинаты в мозговом слое и почечных сосочках |
Примечание: * - суммарная доза для метамизола обычно не менее 500 г, фенацетина - не менее 300 г, диклофенака не менее 150 г.
Использование этих критериев не означает, что ХТИН не может развиться в более короткие сроки и при применении меньших суммарных доз препаратов.
Дифференциальная диагностика
Дифференциальная диагностика осуществляется посиндромно. Ключевым может явиться мочевой синдром, а также синдром АГ или ХПН.
В случае наличия гематурии или протеинурии необходимо исключить ряд заболеваний, проявляющихся данными изменениями мочевого осадка (рис. 2, 3).
Рекомендация 3. Больным с ХБП 3-й стадии, а также хроническими заболеваниями почек в ассоциации с сердечно-сосудистыми заболеваниями или без терапия НПВС противопоказана, за исключением особых ситуаций (когда польза от применения НПВС превышает вред). В случае их назначения следует избегать более высоких доз, чем те, которые обычно рекомендованы (1В). При ХБП 4-5-й стадий терапия НПВС противопоказана, за исключением эпизодического приема по неотложным показаниям (1В).
Лечение
Перед началом лечения НПВС и анальгетиками должны быть оценены факторы риска почечного повреждения и почечная функция. К их числу следует отнести ХБП, сахарный диабет, АГ, хроническую сердечную недостаточность. Крайне нежелательно назначать по два и более препарата, обладающих анальгетической и противовоспалительной активностью. При снижении почечной функции прием НПВС целесообразно прекратить полностью.
Рекомендация 4. Необходимо по возможности установить причинный фактор, длительное воздействие которого вызвало развитие ХТИН, и устранить или ослабить его воздействие на организм (2С). В частности отмена НПВС и анальгетиков сопровождается замедлением прогрессирования ХПН (2С).
В случае известной причины интерстициального нефрита, терапия направлена на ее устранение или уменьшение ее влияния на интерстиций. К этиотропным видам терапии следует отнести отказ от приема анальгетиков, НПВС и других препаратов, вызвавших поражение почек. Следует предположить важную роль блокаторов РААС (ингибиторы АПФ и антагонисты рецепторов ангиотензина II) в замедлении прогрессирования почечной недостаточности вследствие антигипертензивного, антифибротического и противовоспалительного действия. Поскольку наблюдаемые в почечной паренхиме патологические изменения в основном представлены интерстициальным фиброзом, лечение может быть направлено скорее на профилактику его развития, а также на замедление темпов почечного ремоделирования (устранение гиперфильтрации, снижение явлений фиброзирования и асептического воспаления, повышение устойчивости тубулярного аппарата к оксидативному стрессу и т.д.). Возможно, в качестве препаратов выбора должны выступать блокаторы РААС - ингибиторы АПФ и антагонисты рецепторов ангиотензина II, ренопротективная активность которых доказана при целом ряде хронических ренопаренхиматозных заболеваний. Главным аргументом их потенциальной эффективности при НПВС-нефрите является способность замедлять прогрессирование ХПН за счет торможения ремоделирования тубулоинтерстициальной ткани, опосредованного активацией РААС.
Рекомендация 5. Терапия ингибиторами АПФ или блокаторами рецепторов к ангиотензину II может рекомендоваться для лечения АГ на фоне ХТИН наравне с β-блокаторами, диуретиками и блокаторами медленных кальциевых каналов, однако учитывая ее ренопротективный потенциал, она имеет преимущества перед другими классами (2С). Применение ингибиторов АПФ или блокаторов рецепторов к ангиотензину II у нормотензивных пациентов в субгипотензивных дозах с целью ренопротекции может рекомендоваться с осторожностью, не имеет хорошей доказательной базы и лишь основывается на результатах немногочисленных открытых клинических и экспериментальных исследований (НГD).
Рекомендация 6. Необходимо осуществлять коррекцию модифицируемых факторов риска прогрессирования ХПН и смерти больных с ХПН (Аг, гипергликемия, дислипидемия, гиперфосфатемия, гиперурикемия), что будет способствовать замедлению прогрессирования ХПН у больных с ХТИН (1В).
Важно помнить о том, что развившаяся ХПН, вне зависимости от ее происхождения, требует коррекции факторов риска ее прогрессирования. Эта коррекция осуществляется как немедикаментозно (при ХБП 3-4-й стадий - низкобелковая диета с ограничением приема белка до 0,8 г/кг/сут с возможным добавлением в терапию кетокислот), так и медикаментозно (антигипертензивная, гиполипидемическая, фосфор-связывающая, антигиперпаратиреоидная, антигиперурикемическая и др.).
Рекомендация 7. Терапия глюкокортикоидами при ХТИН не проводится, исключение составляют клинические ситуации, при которых показана терапия глюкокортикоидами заболевания, в рамках которого развивается ХТИН (НГD).
Терапия диуретиками применяется при развитии олигурии и гипергидратации. Возможно применение антиагрегантов, улучшающих микроциркуляцию в почках. Данный вид терапии имеет весьма скромную доказательную базу, состоящую преимущественно из открытых клинических исследований и лишь небольшого количества рандомизированных слепых исследований.
Рекомендация 8. Возможно применение в терапии ХТИН препаратов, улучшающих почечную микроциркуляцию (пентоксифиллин, ацетилсалициловая кислота, сулодексид, тиоктовая кислота и др.), что может замедлять темпы прогрессирования ХПН (НГD).
Вероятным позитивным эффектом могут обладать препараты, сделанные на основе простагландинов, обладающие сосудорасширяющими и антиагрегантными свойствами, нивелирующие эффекты НПВС.
Течение и прогноз
У больных с ХТИН примерно в половине случаев регистрируется ХПН. Постепенно наблюдается снижение почечной функции с развитием ХБП 5-й стадии. Прогрессирование ХПН при ХТИН обычно происходит меньшими темпами, чем при хронических гломерулонефритах, однако и методы терапевтического сдерживания при данной патологии весьма ограничены. В случае развития ХБП 5-й стадии больному осуществляется ЗПТ в соответствии с общепринятыми подходами. Поскольку ХТИН чаще развивается в пожилом и старческом возрасте, при ведении больных необходимо учитывать сопутствующую сердечно-сосудистую патологию и сахарный диабет, часто наблюдаемые у пациентов этой возрастной группы.
СПИСОК ЛИТЕРАТУРЫ
-
Балкаров И.М., Лебедева М.В., Щербак А.В., Мухин Н.А. Клиника, диагностика и лечение хронического тубулоинтерстициального нефрита // Клин. фармакол. и тер. 2000. Т. 9, № 5. С. 81-85.
-
Батюшин М.М., Дмитриева О., Терентьев В.П. Роль анальгетиков и нестероидных противовоспалительных препаратов в развитии интерстициальных поражений почек // Нефрология и диализ. 2006. № 3. С. 239-244.
-
Батюшин М.М., Пасечник Д.Г. Протеинурия: вопросы дифференциальной диагностики // Consilium Medicum. 2013. № 7. С. 48-56.
-
Батюшин М.М., Пасечник Д.Г. Гематурия: понятие, причины и основы дифференциальной диагностики // Consilium Medicum. 2012. № 1. С. 12-18.
-
Есаян А.М. Тканевая ренин-ангиотензиновая система почки. Новая стратегия ренопротекции // Нефрология. 2002. Т. 6, № 3. С. 10-14.
-
Шилов Е., Андросова С. Лекарственные поражения почек // Врач. 2002. № 6. С. 47-49.
-
Ayasreh-Fierro N., Ars-Criach E., Lopes-Martin V. et al. Familial chronic interstitial nephropathy with hyperuricaemia caused by the UMOD gene // Nefro-logia. 2013. Vol. 33, N 4. P. 587-592.
-
Akhund L., Quinet R.J., Ishaq S. Celecoxib-related papillary necrosis // Arch. Intern. Med. 2003. Vol. 163. P. 114-115.
-
Bouvy M.L., Heerdink E.R., Hoes A.W., Leufkens H.G. Effects of NSAIDs on the incidence of hospitalisations for renal dysfunction in users of ACE inhibitors // Drug Saf. 2003. Vol. 26, N 13. P. 983-989.
-
Brater D.C. Anti-inflammatory agents and renal function // Semin. Arthritis Rheum. 2002. Vol. 32. P. 33-42.
-
Ekici A.B., Hackenbeck T., Moriniere V. et al. Renal fibrosis is the common feature of autosomal dominant tubulointerstitial kidney diseases caused by mutations in mucin 1 or uromodulin // Kidney Int. 2014. Vol. 86, N 3. P. 589-99.
-
Feinstein A.R., Heinemann L.A., Curhan G.C., Delzell E. et al. Relationship between nonphenacetin combined analgesics and nephropathy: a review. Ad Hoc Committee of the International Study Group on Analgesics and Nephropa-thy // Kidney Int. 2000. Vol. 58, N 6. P. 2259-2264.
-
Galesic K., Morovic-Vergles J., Jelakovic B. Nonsteroidal antirheumatics and the kidney // Reumatizam. 2005. Vol. 52, N 2. P. 61-66.
-
Gooch K., Culleton B.F., Manns B.J. et al. NSAID use and progression of chronic kidney disease // Am. J. Med. 2007. Vol. 120. P. 280.e1-280.e7.
-
Lanas A., Benito P., Alonso J. et al. Safe Prescription Recommendations for Non Steroidal Anti-Inflammatory Drugs: Consensus Document Elaborated by Nominated Experts of Three Scientific Associations (SER-SEC-AEG) // Reuma-tol. Clin. 2014. Vol. 10, N 2. P. 68-84.
-
Leven C., Hudier L., Picard S. et al. Prospective study of drug-induced interstitial nephritis in eleven French nephrology units // Presse Med. 2014. Vol. 43, N 11. P. e369-e376.
-
Liu S., Soong Y., Seshan S.V., Szeto H.H. Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis // Am. J. Physiol. Renal Physiol. 2014. Vol. 306, N 9. P. F970-F980.
-
Nanayakkara S., Senevirathna S.T., Abeysekera T. et al. An integrative study of the genetic, social and environmental determinants of chronic kidney disease characterized by tubulointerstitial damages in the North Central Region of Sri Lanka // J. Occup. Health. 2014. Vol. 56, N 1. P. 28-38.
-
Plantinga L., Grubbs V., Sarkar U. et al. Nonsteroidal anti-inflammatory drug use among persons with chronic kidney disease in the United States // Ann. Fam. Med. 2011. Vol. 9. P. 423-430.
-
Shirali A.C., Perazella M.A. Tubulointerstitial injury associated with chemo-therapeutic agents // Adv. Chronic Kidney Dis. 2014. Vol. 21, N 1. P. 56-63.
ТУБУЛОПАТИИ. ГИПОФОСФАТЕМИЧЕСКИЙ РАХИТ
Раздел 1. Введение, генетика, эпидемиология, патогенез
Рекомендация 1.1. Под тубулопатиями (лат. tubulus трубочка + греч. pathos страдание, болезнь) следует понимать гетерогенную группу патологических состояний, характеризующихся врожденным или приобретенным дефектом канальцевых функций почек (без существенных изменений клубочковой фильтрации) с нарушением обмена веществ, соответствующим характеру дефекта. Большинство тубулопатий имеет наследственный характер и выявляется в детском возрасте. У взрослых тубулопатии также могут представлять собой поздно распознанное генетическое заболевание, но чаще, чем у детей, они обусловлены приобретенным дефектом канальцевых функций при заболевании почек (интерстициальный нефрит, амилоидоз), опухолях и аутоиммунных заболеваниях. Основными в группе тубулопатий являются гипофосфосфатемический рахит, ренальный тубулярный ацидоз, синдром Фанкони, почечная глюкозурия и почечный диабет (НГ).
Рекомендация 1.2. Гипофосфатемический рахит (ГФР) (синонимы: гипофосфатемия, Х-сцепленная; витамин D-резистентный рахит; гипофосфатемический витамин D-резистентный рахит) - наследственное рахитоподобное заболевание, в основе которого лежит ферментный дефект, проявляющийся снижением реабсорбции фосфатов в проксимальных отделах почечных канальцев, приводящим к гиперфосфатурии и гипофосфатемии, характеризуется клинической картиной рахита или остеомаляции, не поддающихся лечению обычными для терапии рахита дозами витамина D (НГ).
Рекомендация 1.3. Генетическая диагностика важна, так как от генетической формы ГФР зависят клинические и биохимические характеристики ГФР, а также терапия и прогноз заболевания (1А).
Суммарная частота ГФР составляет 1:20000 детского населения. Исследования молекулярной природы этого заболевания свидетельствуют о том, что ГФР отличается не только широкой вариабельностью клинических проявлений, но и является генетически гетерогенным заболеванием (одной из форм наследственных гипофосфатемий), что и определяет клинический полиморфизм патологии.
Клинические и биохимические характеристики ГФР, а также лечение в значительной степени зависят от этиологии заболевания.
В настоящее время известны следующие клинико-генетические формы ГФР (табл. 1).
| Клинические формы | Локализация гена | Название гена | Тип наследования |
|---|---|---|---|
ГФР, Х-сцепленный доминантный (OMIM 307800) |
Хромосома Хр22.1-р22.2 |
PHEX (Phosphate Regulating Gene with Homologies to Endopeptidases, X-linked) |
Х-сцепленный доминантный |
ГФР, Х-сцепленный рецессивный (OMIM 300554) |
Хромосома Хр11.22 |
CLCN5 (H/Cl exchange transporter 5) |
Х-сцепленный рецессивный |
ГФР, аутосомно-доминантный (OMIM 193100) |
Хромосома 12р13.3 |
FGF 23 (Fibroblast growth factor 23) |
Аутосомно-доминантный |
ГФР с нарушением дентиногенеза, аутосомно-рецессивный (OMIM 241520) |
Хромосома 4q21 |
DMP1 (dentin matrix protein 1) |
Аутосомно-рецессивный |
ГФР с генерализованной кальцификацией артерий, аутосомно-рецессивный (OMIM 613312) |
Хромосома 6 q22-q23 |
ENPP1 (ecto-nucleotide pyrophosphatase/ phosphodieste-rase 1) |
Аутосомно-рецессивный |
ГФР с гиперкальциурией (OMIM241530) |
Хромосома 9q34 |
SLC34A3 (solute carrier family 34 (sodium phospat, member 3) |
Аутосомно-рецессивный |
ГФР с гиперпаратиреоидизмом (OMIM 612089) |
Хромосома 13q13.1 |
||
Гены Na/P транспортеры: SLC34A3 (OMIM 609826) SLC34A1 (OMIM 182309) |
Хромосома 9q34 5q35 |
Аутосомно-рецессивный Аутосомно-рецессивный |
Наибольший удельный вес в группе ГФР занимает Х-сцепленная доминантная форма, которая встречается в 80% среди всех случаев ГФР.
Ген этой формы ГФР (имеет символы РHEX- ген__ (Phosphate Regulating Gene with Homologies to Endopeptidases, X-linked), HYP, HPDR1, LXHR) локализован на хромосоме Xp22.1-p22.2 и состоит из 22 небольших экзонов.
Молекулярно-генетические исследования показали, что ген РНЕХ кодирует фосфатрегулирующий белок, относящийся к классу эндопептидаз и состоящий из 749 аминокислотных остатков, и имеет молекулярную массу 86,5 кДа. Экспрессия гена РНЕХ происходит в остеобластах, остеоцитах и одонтобластах. Основные функции данного белка заключаются в регулировании реабсорбции фосфатов и метаболизма витамина D. РНЕХ может активировать или инактивировать паракринные или аутокринные факторы, влияющие на минерализацию костной и зубной ткани, а также на циркулирующие факторы, регулирующие реабсорбцию фосфатов и метаболизм витамина D. Ген контролирует активность N/Р переносящего белка в почечных канальцах и эпителии кишечника. Генетический дефект приводит к нарушению реабсорбции фосфатов в канальцах почек и его всасыванию в тонкой кишке. Однако последние данные свидетельствуют о том, что в основе патогенеза Х-сцепленного ГФР также лежит избыточное действие фактора роста фибробластов-23 (FGF23).
На сегодняшний день описано более 250 мутаций РНЕХ-гена, которые представлены различными классами, в том числе нонсенс-мутации (17-19%), миссенс-мутации (21-22%), делеции (24-30%), инсерции (11-12%), мутации, ведущие к нарушению сплайсинга (18-24%) и др.
Фенотипические признаки Х-сцепленного доминантного ГФР сильно варьируют от изолированной гипофосфатемии до выраженной деформации нижних конечностей.
Манифестация Х-сцепленного ГФР чаще всего наступает на втором году жизни. Ведущими признаками являются рахитоподобные изменения скелета, преимущественно нижних конечностей, деформации могут быть не только варусными, но и вальгусными. Эти изменения сопровождаются задержкой физического развития и нарушением походки детей («утиная походка»). Поражения скелета носят прогрессирующий характер и способствуют задержке статико-моторных функций ребенка.
Рентгенологические изменения костей выявляются через 3-4 мес после манифестации заболевания: генерализованный остеопороз, увеличение метафизов, метафизарные поверхности имеют неровные контуры, по мере прогрессирования заболевания появляются и более глубокие изменения.
Характерными биохимическими признаками заболевания являются: гипофосфатемия, повышение активности щелочной фосфатазы в 1,52 раза, нормальный уровень кальция и паратгормона в сыворотке крови, низкий уровень кальцитриола (1,25(ОН)2D3), гиперфосфатурия.
ГФР, Х-сцепленный рецессивный обусловлен мутациями в гене CLCN5, локализованном на хромосоме Х в локусе - Хр11.22 и имеет 12 экзонов.
В результате мутаций снижается реабсорбция кальция и фосфора в почечных канальцах, развиваются нефрокальциноз и камнеобразование в почках, прогрессирующая ПН. Описано около 148 мутаций CLCN5- гена, которые представлены различными классами, в том числе нонсенс-мутациями, миссенс-мутациями, делециями, мутациями, ведущими к нарушению сплайсинга и др. Заболевание встречается только у мальчиков.
Клинические признаки сходны с Х-сцепленной доминантной формой заболевания и аутосомно-доминантным вариантом заболевания.
ГФР, аутосомно-доминантный встречается редко и обусловлен мутацией в гене, кодирующем фактор роста фибробластов 23 (FGF23), локализованном на хромосоме 12 в локусе 12р13.3. FGF23 - белок с молекулярной массой 30 кДа, состоящий из 251 аминокислотного остатка. Фактор роста фибробластов-23 (FGF23) является новым недавно открытым циркулирующим пептидом, регулирующим метаболизм фосфора и витамина D. Ген экспрессируется в почках, в меньшей степени в мозге, тимусе, тонком кишечнике, сердце, печени, лимфатических узлах, щитовидной и паращитовидной железах, костном мозге и в небольших количествах в опухолях при онкогенной остеомаляции. Свое действие FGF23 осуществляет через специфические рецепторы FGF (FGFRs), расположенные в проксимальных канальцах почек (FGFR1, FGFR3 и FGFR4). FGF23 самостоятельно не может связываться с FGF-рецептором. Для реализации эффектов FGF23 на органы необходим белок Klotho, который присоединяется к рецептору FGF23 и С-терминалу этого гормона. Белок Klotho является трансмембранным протеином и локализуется в дистальных извитых канальцах, паращитовидных железах, сосудистом сплетении, гипофизе и в репродуктивных органах. Активация FGFR приводит к активации внутриклеточных сигнальных путей, которые, в свою очередь, уменьшают экспрессию NaPi-2a, NaPi-2c и 1а-гидроксилазы, что приводит к снижению реабсорбции фосфора в проксимальных канальцах и снижению синтеза 1,25(OH)2D3. FGF23 оказывает фосфатурический эффект, стимулирует секрецию паратгормона и тормозит 1а-гидроксилазную активность почек, приводя к снижению синтеза кальцитриола и его уровня в сыворотке крови.
При аутосомно-доминантной форме заболевания генетический риск для сибсов пробанда составляет 50% независимо от пола потомства. Заболевание может наблюдаться как у мальчиков, так и у девочек.
Клиническая картина при данной форме ГФР зависит от возраста дебюта заболевания. При манифестации заболевания в подростковом и более старшем возрасте отмечаются боли в ногах, слабость, повышенная утомляемость, псевдопереломы либо спонтанные переломы, остеомаляция, но деформации нижних конечностей отсутствуют. При дебюте заболевания в детском возрасте (на 1-3-м году жизни, чаще на 2-м году) клиническая картина сходна с Х-сцепленным ГФР. Клинические проявления характеризуются нарушением походки, незначительной степенью костных деформаций, чаще всего по варусному типу, мало отражающихся на задержке роста. Костные деформации захватывают преимущественно нижние конечности. Задержка роста умеренная. Характерная особенность - развитие пародонтоза и дентальных абсцессов. Метаболические расстройства проявляются умеренной гипофосфатемией и гиперфосфатурией, нормальными показателями уровня кальция крови и незначительным повышением щелочной фосфатазы сыворотки крови. В некоторых случаях в постпубертатном периоде может возникать спонтанная ремиссия в виде нормализации биохимических показателей.
ГФР с нарушением дентиногенеза, аутосомно-рецессивный является редкой формой и обусловлен инактивирующей мутацией в аутосомном гене - DMP1 (dentin matrix protein 1), локализованном на хромосоме 4 в локусе 4q21. DMP1, первоначально названный как AG1, является кислым неколлагеновым фосфопротеином и относится к семейству SIBLING (small integrin bindin gligand N-glycated), включающему в себя остеопонтин, костный сиалопротеин, DMP1, дентин сиалопротеин (DSPP), энамелин и внеклеточный фосфогликопротеин (MEPE), которые являются неколлагеновыми белками внеклеточного матрикса костной ткани. Первоначально DMP1 был обнаружен в дентине, затем в костной и хрящевой тканях; недавно был обнаружен в печени, мышцах, поджелудочной железе, почках, головном мозге, слюнных железах. DMP1 экспрессируется в остеобластах и остеоцитах и состоит из 531 аминокислотного остатка. DMP1 имеет несколько функций в регуляции постнатальной минерализации костной ткани. Этот белок играет важную роль в пролиферации остеоцитов, а также в подавлении FGF23.
Заболевание может наблюдаться как у мальчиков, так и у девочек. Клиническая картина данного варианта ГФР сходна с Х-сцепленной доминантной и аутосомно-доминантной формой: отмечается гиперфосфатурия, гипофосфатемия, очень низкий уровень 1,25(OH)2D3, повышение активности щелочной фосфатазы, деформация нижних конечностей, обязательным симптомом является поражение зубов в виде дистрофии эмали.
ГФР c генерализованной кальцификацией артерий, аутосомно-рецессивный обусловлен инактивирующей мутацией в гене ENPP1 (ecto-nucleotide pyrophosphatase/phosphodiesterase 1), локализованном на хромосоме 6 в локусе 6q22-q23 и имеет 25 экзонов. Ген ENPP1 кодирует белок, представляющий собой трансмембранный гликопротеин II типа - NPP1 (ecto-nucleotide pyrophosphatase/phosphodiesterase 1). NPP1 регулирует кальцификацию мягких тканей, минерализацию костей и суставных хрящей путем генерации внеклеточного неорганического фосфора, который является важным физиологическим ингибитором кальцификации.
Заболевание может наблюдаться как у мальчиков, так и у девочек. Клинические проявления данного варианта сходны с классическим ГФР, однако заболевание сопровождается кальцификацией артерий (аорты, крупных и средних артерий).
ГФР c гиперкальциурией, аутосомно-рецессивный обусловлен мутацией в гене SLC34A3, локализованном на хромосоме 9 в локусе 9q34, кодирующем NaPi-IIc, и отличается от других форм ГФР тем, что заболевание проявляется не только гипофосфатемией и гиперфосфатурией, а также гиперкальциурией в связи с увеличением в сыворотке крови 1,25-дигидроксихолекальциферола и повышением абсорбции кальция в кишечнике, в то время как в крови уровень Са в норме; уровень парат-гормона остается в пределах нормы. Заболевание манифестирует в раннем возрасте и характеризуется рахитическими деформациями нижних конечностей, задержкой роста, повышенным ренальным клиренсом фосфатов и гиперкальциурией, в то время как концентрация кальция в сыворотке крови остается нормальной. Заболевание может наблюдаться как у мальчиков, так и у девочек.
ГФР с гиперпаратиреоидизмом связано с мутацией в гене, локализованном на хромосоме 13, в локусе 13q13.1. Молекулярно-генетические исследования этого варианта заболевания пока еще не дали окончательного ответа на вопрос о типе наследственной передачи заболевания и механизмах его развития. Предполагается аутосомно-рецессивное наследование. Заболевание может наблюдаться как у мальчиков, так и у девочек.
Клинические проявления данного варианта сходны с Х-сцепленным доминантным ГФР, однако заболевание сопровождается гиперпаратиреоидизмом и поздним развитием гиперкальциемии.
Раздел 2. Клиническая диагностика
Рекомендация 2.1. Клиническая диагностика ГФР должна включать сбор данных о наличии рахитических изменений у кровных родственников (НГ). При Х-сцепленных формах заболевания рекомендуется исследование уровня фосфатов крови у матерей.
Рекомендация 2.2. Клиническими критериями диагностики являются сроки начала заболевания, появление рахитоподобных изменений скелета, преимущественно нижних конечностей (варусные или вальгусные деформации), задержка физического развития, нарушение походки детей (утиная походка), прогрессирующий характер поражения скелета (НГ).
Рекомендация 2.3. Рентгенологическими критериями диагностики являются развитие генерализованного остеопороза, увеличение метафизов, метафизарные поверхности имеют неровные контуры (НГ).
Рекомендация 2.4. Биохимическими критериями диагностики являются: гипофосфатемия, повышенная активность щелочной фосфатазы в 1,52 раза, нормальный уровень кальция и паратгормона в сыворотке крови, низкий уровень кальцитриола (1,25(ОН)2D3), гиперфосфатурия (НГ).
Рекомендация 2.5. Дополнительными критериями являются: генерализованная кальцификация артерий, гиперкальциурия, гиперпаратиреоидизм (НГ).
Рекомендация 2.6. Генетическую диагностику следует считать целесообразной в случае нетипичной картины заболевания и при выявлении дополнительных критериев диагностики; генетические исследования следует назначить всем членам семьи, что позволяет выявить доклинические или малосимптомные случаи заболевания и определить тип наследования, что важно для генетического прогноза семьи; проведение популяционного генетического скрининга нецелесообразно (НГ).
Раздел 3. Лечение
Рекомендация 3.1. Методы этиологического (генотерапия) лечения ГФР не разработаны; основой лечения ГФР является патогенетическая и симптоматическая терапия, направленная на:
Рекомендация 3.2. Назначение медикаментозных средств должно быть строго индивидуальным в зависимости от формы, характера и степени изменения клинических и биохимических параметров после комплексного обследования (НГ).
Рекомендация 3.3. Лечение в период активного роста детей частично устраняет скелетные деформации, уменьшает количество необходимых операций, а также увеличивает конечный рост детей. Ранняя верификация заболевания и раннее начало лечения позволяют оптимизировать эффект от терапии (НГ).
Рекомендация 3.4. При наличии тяжелых костных деформаций, затрудняющих передвижение больных, может быть рекомендовано хирургическое лечение (НГ).
Комментарии. Патогенетическая терапия. Дефицит витамина D. С целью ликвидации дефицита фосфора в крови и уменьшения их потерь с мочой используются витамин D, его активные метаболиты. Базисными препаратами является витамин D и его синтетические аналоги. Ориентировочные дозы препаратов витамина D: для детей 0-3 лет составляют 7000-6000 ЕД/кг/сут, для детей 4-7 лет 6500-4000 ЕД/кг/сут. Начальные дозы витамина D составляют 10000-15 000 ЕД/сут. Повышение доз должно осуществляться под индивидуальным контролем за содержанием кальция и неорганических фосфатов в сыворотке крови и моче, активности щелочной фосфатазы крови, исследование которых должно проводиться каждые 10-14 дней. Из метаболитов витамина D используется отечественный препарат альфакальцидол (Оксидевит♠) в суточной дозе 0,25-3 мкг, а также кальцидол,Ы альфакальцидол (Альфа Д3-Тева♠), кальцитриол. Кальцитриол увеличивает абсорбцию фосфора в кишечнике, однако высокие дозы кальцитриола могут приводить к повышению уровня FGF23 в сыворотке крови.
Симптоматическая терапия. Дефицит фосфора и кальция. Для восполнения дефицита фосфора и кальция в комплекс лечения обязательно включаются препараты фосфора (неорганические фосфаты до 2,0 г в сутки, кальция глицерофосфат 0,5-1 г/сут и др.) и кальция (кальция глюконат или кальция хлорид 1,5-2,0 г/сут). За рубежом для коррекции дефицита фосфора используются препараты, содержащие неорганические фосфаты натрия или калия (редукта, фосфонейрос и др.), которые пока не имеют регистрации в России. В комплекс лечения детей с ГФР включается Остеогенон♠ - оссеин гидроксиапатитный комплекс. Препарат содержит 178 мг кальция и 82 мг фосфора. В состав препарата входят также неколлагеновые белки - 75 мг и коллагеновые белки - 216 мг. Остеогенон♠ стимулирует остеогенез, ингибирует костную резорбцию, оптимизирует обмен фосфатов. Применяется по 1-2 таблетки 2-3 раза в день.
Отставание в росте. Для ускорения темпов роста больных детей могут применяться препараты гормона роста - соматотропины в виде курсов по 3-4 мес (1 МЕ/кг/неделю). В комплекс лекарственных средств рекомендуется включение бисфосфонатов, в том числе отечественных [этидроновая кислота (Ксидифон♠)].
Осложнения терапии. Лечение проводится под регулярным контролем уровня фосфора и кальция в крови, так как избыток фосфатов может привести к вторичному гиперпаратиреозу с усилением деминерализации кости, а избыток кальцитриола и альфакальцидола (Оксидевита♠) - к гиперкальциурии и нефрокальцинозу.
Хирургическое лечение костных деформаций. Хирургическое лечение показано пациентам при наличии тяжелых костных деформаций, затрудняющих передвижение больных. Оптимальными сроками для успешного проведения хирургической коррекции костных деформаций является возраст 6-8 лет. Обязательным условием для проведения хирургического лечения является достижение стойкой клинико-лабораторной ремиссии в течение не менее 2 лет.
Раздел 4. Перспективы в терапии гипофосфатемического рахита
Одним из перспективных направлений, которые находятся сейчас в стадии клинических разработок и которые, возможно, приведут к формулировке соответствующих рекомендаций, является использование принципиально нового препарата цинакалцет (Мимпара♠), оказывающего антигиперпаратиреоидное действие за счет аллостерической модуляции кальцийчувствительных рецепторов, расположенных на поверхности главных клеток околощитовидных желез, и снижающего порог их реакции на внеклеточный кальций, что обеспечивает контроль секреции паратгормона и обратное развитие гиперплазии паращитовидных желез у больных ГФР с гиперпаратиреозом. По имеющимся данным, у пациентов с ХПН и гиперпаратиреозом улучшаются показатели фосфорно-кальциевого гомеостаза, повышается плотность костной ткани, и препарат предупреждает развитие кальциноза сердца и сосудов.
Реальных перспектив разработки этиотропной терапии ГРФ в настоящее время нет.
СПИСОК ЛИТЕРАТУРЫ
-
Волгина Г.В., Балкарова О.В., Штандель В.С., Ловчинский Е.В. Кальцимиметики - новый этап в лечении гиперпаратиреоза // Леч. врач. 2011. № 3. С. 1-4.
-
Мальцев С.В., Архипова Н.Н. Генетически детерминированные нарушения обмена фосфатов у детей и пути их коррекции // Казан. мед. журн. 2004. Т. 85, № 5. С. 374-376.
-
Молчанова М.С., Петросян Э.К., Панкратенко Т.У. и соавт. Опыт применения цинакалцета у детей с хронической болезнью почек стадии // Клин. нефрология. 2011. № 6. С. 66-70.
-
Мусаева А.В. Катамнез детей и подростков с витамин^-резистентным гипофосфатемическим рахитом : автореф. дис. … канд. мед. наук. СПб., 2012. 30 с.
-
Новиков П.В. Рахит и наследственные рахитоподобные заболевания у детей. М. : Триада-Х, 2006. С. 194-224.
-
Новиков П.В. Лечебная тактика коррекции метаболических расстройств у детей с наследственными заболеваниями обмена веществ. М. : Оверлей, 2011. С. 104-132.
-
Юрьева Э.А., Вельтищев Ю.Е., Игнатова М.С. Тубулопатии // Детская нефрология: руководство для врачей / под ред. М.С. Игнатовой. 3-е изд. М. : Медицинское информационное агентство, 2011. С. 355-389.
-
Bergwitz C., Roslin N.M., Tieder M. et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeo-stasis // Am. J. Hum. Genet. 2006. Vol. 78, N 2. P. 179-192.
-
Carpenter T.O. The expanding family of hypophosphatemic syndromes // J. Bone Miner. Metab. 2012. Vol. 30, N 1. P. 1-9.
-
Carpenter Imel E.A., Holm I.A. et al. A clinician’s guide to X-linked hypophosphatemia // J. Bone Miner. Res. 2011. Vol. 26, N 7. P. 1381-1388.
-
Devuyst O., Thakker R.V. Dent’s disease // Orphanet J. Rare Dis. 2010. Vol. 5. P. 28.
-
Farrow E.G., Davis S.I., Ward L.M. et al. Molecular analysis of DMP1 mutants causing autosomal recessive hypophosphatemic rickets // Вone. 2009. Vol. 44. P. 287-294.
-
Gambaro G., Vezzoli G., Casari G. et al. Genetics of hypercalciuria and calcium nephrolithiasis: from the rare monogenic to the common polygenic forms // Am. J. Kidney Dis. 2004 Dec. Vol. 44, N 6. P. 963-986.
-
Gattineni J., Baum M. Genetic disorders of phosphate regulation // Pediatr. Nephrol. 2012. Vol. 27, N 9. P. 1477-1487.
-
Gattineni J., Baum M. Regulation of phosphate transport by fibroblast growth factor 23 (FGF23): implications for disorders of phosphate metabolism // Pediatr. Nephrol. 2010. Vol. 25, N 4. P. 591-601.
-
Gaucher C., Walrant-Debray O., Nguyen T.M. et al. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets // Hum. Genet. 2009. Vol. 125, N 4. P. 401-411.
-
Ichii M., Ishimura E., Okuno S. et al. Decreases in parathyroid gland volume after cinacalcet treatment in hemodialysis patients with secondary hyperparathyre-oidism // Nephron. Clin. Pract. 2010. Vol. 115, N 3. P. 195-202.
-
Levy-Litan V., Hershkovitz E., Avizov L. et al. Autosomal-recessive hypo-phosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene // Am. J. Hum. Genet. 2010. Vol. 8, N 6. P. 273-278.
-
Lindberg J.S., Culleton B., Wong G. et al. Cinacalcet HCl , an oral calcimi-metic agent for the treatment of secondary hyperparathyreoidism in hemodialysis and peritoneal dislysis: a randomized double-blind multicenter study // J. Am. Soc. Nephrol. 2005. Vol. 16, N 3. P. 800-807.
-
Ling Y., Rios H.F., Myers E.R. et al. DMP1 depletion decreases bone mineralization in vivo: an FTIR imaging analysis // J. Bone Miner. Res. 2005. Vol. 20, N 12. P. 2169-2177.
-
Morey M., Castro-Feijoo L., Barreiro J. et al. Genetic diagnosis of X-linked dominant hypophosphatemic rickets in a cohort study: Tubular reabsorption of phosphate and 1,25(OH)2D serum levels are associated with PHEX mutation type // BMC Med. Genet. 2011. Vol. 12. P. 116.
-
Nitschke Y., Baujat G., Botschen U. et al. Generalized arterial calcification of infancy and pseudoxanthomaelasticum can be caused by mutations in either ENPP1 or ABCC6 // Am. J. Hum. Genet. 2012. Vol. 90, N 1. P. 25-39.
-
Perwad F., Azam N., Zhang M.Y. et al. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice // Endocrinology. 2005. Vol. 146, N 5. P. 358-5364.
-
Rutsch F., Boyer P., Nitschke Y. et al. Hypophosphatemia, hyperphospha-turia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy // Circ. Cardiovasc. Genet. 2008. Vol. 1, N 2. P. 133-140.
-
Velayoudom-Cephise F.L., Vantyghem M.C., Wemeau J.L. Hereditary hypophosphatemia in adults // Presse Med. 2005. Vol. 34, N 22. P. 1720-1726.