Нефрология. Клинические рекомендации / под ред. Е. М. Шилова, А. В. Смирнова, Н. Л. Козловской. - М. : ГЭОТАР-Медиа, 2016. - 816 с. - ISBN 978-5-9704-3714-8. |
Аннотация
Первые национальные клинические рекомендации по нефрологии подготовлены коллективом экспертов, в который вошли не только специалисты-нефрологи ведущих нефрологических школ России, но и представители других медицинских специальностей, тесно сотрудничающие с нефрологами, - кардиологи, эндокринологи, инфекционисты, педиатры, генетики. Издание содержит информацию по наиболее распространенным нефрологическим заболеваниям и синдромам. Представленные в нем клинические рекомендации детально описывают действия врача по диагностике, лечению, профилактике и реабилитации пациентов. Соблюдение международной методологии при подготовке клинических рекомендаций гарантирует их современность, достоверность, обобщение лучшего мирового опыта и знаний, обеспечивает возможность практического применения. Именно поэтому клинические рекомендации обладают преимуществами по сравнению с традиционными источниками информации (учебники, руководства, монографии), что позволит врачу в короткие сроки принимать обоснованные решения в сложных клинических ситуациях. Клинические рекомендации по нефрологии предназначены не только практикующим врачам-нефрологам, но и терапевтам, педиатрам, представителям смежных дисциплин. Они также могут использоваться для обучения студентов старших курсов и клинических ординаторов терапевтических специальностей.
1. Определение. Терминология
Рекомендация 1.1. Мембранопролиферативный гломерулонефрит (МБПГН) - это генерический термин (морфологический синдром), объединяющий группу гломерулопатий, имеющих сходную морфологическую картину при световой микроскопии биоптатов, но различающихся по этиологии, патогенезу, иммуногистохимическим и ультраструктурным (электронная микроскопия) изменениям почечной паренхимы (НГ).
Комментарий. В настоящее время достигнуты значительные успехи в понимании этиологии и особенно патогенеза МБПГН, что позволяет рассматривать данную морфологическую форму как весьма неоднородную группу заболеваний. Сохранились прежние представления о клиническом подразделении МБПГН на идиопатическую (с неизвестной этиологией) и вторичные формы, причем последние преобладают. В связи с этим данные прошлых лет о распространенности МБПГН в популяции следует воспринимать с осторожностью. По данным крупных морфологических регистров в странах Западной Европы, распространенность МБПГН варьирует от 4,6 до 11,3%, а в США не превышает 1,2%, составляя примерно 1-6 человек на 1 млн населения. Напротив, в странах Восточной Европы, Африки и Азии распространенность МБПГН, по некоторым данным, достигает 30%, что связывают с большей распространенностью инфекций, прежде всего вирусных гепатитов В и С. Активные меры профилактики инфекций, по-видимому, объясняют наметившуюся в последние 15-20 лет явную тенденцию к снижению распространенности МБПГН в большинстве регионов мира, тем не менее МБПГН остается 3-й и 4-й по счету причиной ТПН среди всех остальных форм первичного ГН.
Синонимами термина «мембранопролиферативный гломерулонефрит» являются «мезангиокапиллярный гломерулонефрит (МКГН)», а в отечественной литературе - «мембранознопролиферативный гломерулонефрит». Предпочтительным следует считать термин «мембранопролиферативный гломерулонефрит».
2. Клиническая презентация
Рекомендация 2.1. Клиническая презентация МБПГН (почечныесиндромы) идентична при идиопатическом (с неизвестной этиологией) и вторичном вариантах заболевания (1В).
Рекомендация 2.2. По характеру клинической картины невозможно предсказать морфологический тип МБПГН (1В).
Рекомендация 2.3. Клиническая дифференциальная диагностика МБПГН должна изначально базироваться на полном и достоверном исключении всех возможных вторичных причин (табл. 1) (НГ).
Комментарий. Несмотря на патогенетическую и морфологическую гетерогенность МБПГН, клиническая презентация со стороны почек идентична. У половины больных в анамнезе отмечаются указания на недавно (до одной недели) перенесенную инфекцию верхних дыхательных путей. В ряде случаев выявляется клинический феномен - синфарингитическая макрогематурия, что заставляет проводить дифференциальную диагностику с IgA-нефропатией. Среди клинических симптомов превалируют: АГ, которая в дебюте отмечается более чем у 30% пациентов, но со временем развивается практически у всех больных, иногда приобретая злокачественное течение; макро- и микрогематурия (практически у 100%); высокая ПУ (нефротическая); прогрессирующее снижение СКФ. Ведущий клинический синдром в дебюте заболевания в 20-30% случаев представлен острым или быстропрогрессирующим НС (БПНС). В первом случае возникает необходимость дифференциальной диагностики с острым постстрептококковым ГН, тем более что в 20-40% случаев МБПГН оказывается высоким титр антистрептолизина-О (АСЛ-О), во втором случае проводят дифференциальную диагностику с анти-ГБМ - нефритом, АНЦА-ассоциированными васкулитами и ТМА. У 40-70% пациентов с самого начала развивается НС [если его нет, то у большинства больных он появляется позже, в 10-20% случаев отмечается рецидивирующая макрогематурия (чаще синфарингитическая)]. Однако у 20-30% больных удается зарегистрировать (как правило, случайно) только изменения в общем анализе мочи в виде сочетания ПУ с микрогематурией и цилиндрурией (изолированный мочевой синдром).
У всех пациентов с острым НС, БПНС и в 50% случаев при других вариантах клинической презентации отмечается снижение СКФ (при БПНС - прогрессирующее) и выявляются многообразные нарушения тубулярных функций (снижение концентрационной способности почек, аминоацидурия, глюкозурия, гиперкалиемия и др.). По клинической картине поражения почек невозможно предсказать тип МБПГН или высказаться определенно о его причине. Чаще (до 80% всех случаев) диагностируется иммуноглобулинпозитивный МБПГН I типа, которым болеют люди любого возраста и пола. Иммуноглобулинпозитивный вариант МБПГН III типа выявляется реже (5-10%). В настоящее время среди нефрологов существует консенсус в отношении идиопатического, иммуноглобулинпозитивного МБПГН I типа (реже III типа), диагноз которого может быть установлен только после исключения вторичных причин (табл. 1). В клинической картине С3-негативной гломерулопатии, как правило, в дебюте превалируют клинико-лабораторные симптомы основного заболевания в сочетании с острым повреждением почек, чаще всего в форме БПНС. Только по истечении острого периода присоединяются высокая ПУ, микрогематурия или формируется НС. Клиническая диагностика болезни плотных депозитов (БПД) облегчается, если помимо почечных синдромов выявляются ассоциированные состояния в виде приобретенной частичной липодистрофии и/или макулярной дистрофии сетчатки глаза (см. ниже).
3. Морфологическая и иммуноморфологическая дифференциальная диагностика
Рекомендация 3.1. Для диагностики МБПГН в соответствии с мировыми стандартами необходимо сочетание нескольких методов морфологического исследования прижизненных биоптатов почечной ткани: световой микроскопии, иммуноморфологии, ультраструктурного анализа (трансмиссионной электронной микроскопии) (НГ).
Рекомендация 3.2. Для проведения светооптического исследования нефробиоптатов необходимо проводить следующие окраски на парафиновых срезах: гематоксилином и эозином, трихромальная окраска по Массону, ПАС-реакция, Конгорот, окраска на эластические волокна и фибрин (AFOG) (1А).
Рекомендация 3.3. Для иммуноморфологического исследования необходимо использовать следующие АТ для выявления диагностически значимых эпитопов: IgA, IgM, IgG, легкие цепи (ЛЦ) λ, κ и фибриноген, фракции комплемента C3, C1q, C2 и С4 (2В).
Рекомендация 3.4. На основании данных ультраструктурного анализа (электронной микроскопии) следует различать МБПГН I типа, БПД и МБПГН III типа (1А).
Рекомендация 3.5. Морфологическую дифференциальную диагностику МБПГН проводят на основании данных иммуноморфологии и электронной микроскопии (1А).
Рекомендация 3.6. Результатом морфологической дифференциальной диагностики должно стать установление следующих патогенетических вариантов МБПГН: иммуноглобулинпозитивный, С3-позитивный МБПГН I или III типов, иммуноглобулиннегативный, С3-позитивный МБПГН I или III типов и болезнь плотных депозитов, иммуноглобулин- и С3-негативный МБПГН (1А).
Рекомендация 3.7. При проведении иммуноморфологического исследования необходимо считать диагностически значимой интенсивность отложения продукта реакции на иммуноглобулины классов A, M, G в структурах гломерул >2+ как при флуоресцентной, так и при светооптической (в проходящем свете) микроскопиях (иммуноглобулинпозитивный вариант МБПГН). Остальные варианты интенсивности отложения продукта реакции на иммуноглобулины (менее 2+) следует считать негативными (иммуноглобулиннегативный вариант МБПГН) (2В).
Рекомендация 3.8. При проведении иммуноморфологического исследования необходимо считать диагностически значимым интенсивность отложения продукта реакции на С3-фракцию комплемента в структурах гломерул ≥2+ как при флуоресцентной, так и при светооптической (в проходящем свете) микроскопиях (С3-позитивный вариант МБПГН). Остальные варианты интенсивности отложения продукта реакции на иммуноглобулины (менее 2+) следует считать негативными (С3-негативный вариант МБПГН) (2В).
Рекомендация 3.9. При отсутствии возможности проведения ультраструктурного анализа (электронной микроскопии) морфологический диагноз должен быть сформулирован на основании данных световой микроскопии и иммуноморфологии (2В).
Рекомендация 3.10. По данным световой микроскопии и иммуноморфологии, следует различать 3 варианта МБПГН (2В):
Рекомендация 3.11. Термин С3-гломерулопатия означает иммуноглобулиннегативный и С3-позитивный МБПГН, включающий 2 формы МБПГН, которые при дальнейшем ультраструктурном анализе могут быть уточнены как иммуноглобулиннегативный, С3-позитивный МБПГН I или III типа или БПД (1А).
Комментарий. Главные морфологические признаки при световой микроскопии представлены пролиферацией клеток и основного вещества мезангиума и утолщением стенок капилляров (базальных мембран), которые часто подвергаются псевдорасщеплению с образованием двухконтурных базальных мембран (феномен «трамвайной линии»). Механизм образования второй базальной мембраны связывают с интерпозицией (внедрением) отростков мезангиоцитов в субэндотелиальное пространство, где они в кооперации с эндотелиоцитами продуцируют новое основное вещество второй расположенной внутри интракапиллярной мембраны.
Помимо пролиферации резидентных клеток отмечается инфильтрация клубочков нейтрофилами и макрофагами (экссудативный компонент воспалительной реакции). Важно отметить, что степень выраженности пролиферативных и экссудативных изменений может варьировать от случая к случаю. Так, в некоторых наблюдениях указанные изменения могут носить фокальный характер (т.е. часть клубочков может оставаться интактной). Полагают, что в этом случае можно говорить о дебюте заболевания. В других наблюдениях, отмечаемых чаще всего, морфологические изменения носят диффузный характер. Описаны также случаи регресса диффузных изменений в фокальные, например, при ликвидации вторичной причины гломерулопатии. В 10% всех случаев МБПГН могут регистрироваться полулуния более чем в 50% клубочков, как отображение выраженности активности пролиферативно-экссудативной реакции. Как правило, в этом случае клинически отмечается быстропрогрессирующий нефритический синдром (БПНС).
Выраженные пролиферативные изменения в мезангиуме очень часто ведут к разделению петель капилляров клубочков на отдельные пучки (лобулы), придающие гломеруле дольчатую структуру. Ранее подобные изменения классифицировали как особую форму МБПГН - лобулярную. В наши дни лобуляция гломерул считается одним из вариантов течения патологического процесса, отражающего степень выраженности пролиферативной реакции и, возможно, ассоциирующегося с длительностью течения МБПГН. По мере дальнейшего прогрессирования, зоны гиперцеллюлярности мезангиума замещаются матриксом, и развивается склероз клубочка. В этой стадии патоморфологические изменения могут имитировать нодулярный диабетический гломерулосклероз. Изменения в сосудах отражают длительность и тяжесть течения АГ. Морфологические изменения клеток канальцев и интерстиция обычно выражены значительно, как правило, они не коррелируют с гломерулярными поражениями, но ассоциируются в клинике с дисфункцией почек. Более детальная характеристика морфологических изменений при МБПГН возможна только при ультраструктурном анализе, который позволяет выделить 3 типа МБПГН. При I типе МБПГН при электронной микроскопии выявляются субэндотелиальные и мезангиальные депозиты. При МБПГН II типа отмечаются интрамембранозные электронноплотные депозиты, которые могут придавать мембране вид «связки колбасы», также присутствуют и мезангиальные депозиты. При МБПГН III типа, помимо субэндотелиальных, регистрируются субэпителиальные (субподоцитарные) депозиты (подтип Burkholder’a), в ряде случаев на базальной мембране формируются выросты около субэпителиальных депозитов (морфологическая картина напоминает МН), сочетающиеся с наличием интрамембранозных отложений (как при II типе МБПГН). Последние придают lamina densa неровный вид (подтип Strife’a и Anders’a).
Подчеркнем, что при световой микроскопии отсутствуют какие-либо типичные морфологические признаки, позволявшие бы прогнозировать диагностику одного из трех типов МБПГН при электронной микроскопии. Более того, при БПД только в 25% случаев при световой микроскопии выявляются типичные признаки МБПГН (описанные выше); в 44% диагностируется мезангиальнопролиферативный ГН, в 17% ГН с полулуниями, в 11% - острый экссудативно-пролиферативный ГН, а в 3% случаев морфологические признаки не поддаются классификации.
Многие исследователи также обращают внимание на существование множества переходных типов при электронной микроскопии, означающих, что даже ультраструктурный анализ не гарантирует окончательного диагноза. Именно поэтому в основу современной классификации МБПГН были положены сведения об иммунопатогенезе, о которых можно судить по данным иммуноморфологии (иммуногистохимии) срезов биоптатов почки.
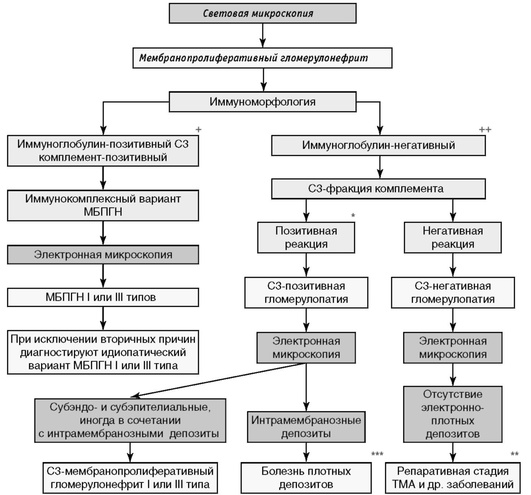
На основании анализа отложений (депозитов) в биоптате почки иммуноглобулинов и фракций комплемента выделяют иммуноглобулин-позитивный и иммуноглобулиннегативный МБПГН (рис. 1).
Наличие иммуноглобулинов и С3-фракции комплемента указывает на иммунокомплексный вариант МБПГН, который характеризуется активацией системы комплемента по классическому пути. Вследствие этого помимо глобулинов и С3-фракции комплемента в почечном биоптате выявляют фракции комплемента Clq, С2, С4, характерные для классического пути активации комплемента. При иммуноглобулиннегативном МБПГН, выявление положительной реакции на С3-фракцию комплемента при отсутствии фракций Clq, С2, С4 будет указывать на активацию комплемента по альтернативному пути. Уже на основании этих данных возможно сформулировать предварительный диагноз С3-позитивной гломерулопатии или С3-гломерулопатии, который далее с помощью электронной микроскопии может быть уточнен как С3-МБПГН I или III типа или БПД (рис. 1).
Учитывая то обстоятельство, что при БПД светооптическая морфологическая картина может не включать характерных для МБПГН признаков (см. выше), допускается постановка диагноза С3-гломерулопатии, однако, подчеркнем еще раз, что при этом должны отсутствовать отложения иммуноглобулинов, Clq- и С4-фракций комплемента, а интенсивность отложения продукта реакции на С3-фракцию комплемента должна составлять не менее 2+. Отсутствие иммуноглобулинов при иммуноморфологическом исследовании и негативная реакция на С3-фракцию комплемента (менее 2+) позволит диагностировать С3-негативную гломерулопатию.
4. Клинико-патогенетическая и лабораторная диагностика
Рекомендация 4.1. Под термином идиопатический МБПГН следует подразумевать иммуноглобулин- и С3-комплементпозитивный вариант МБПГН I или III типов неизвестной этиологии (1А).
Рекомендация 4.2. Иммуноглобулиннегативный, С3-позитивный МБПГН I или III типа и БПД обусловлены наследственными или приобретенными нарушениями в системе альтернативного пути активации комплемента (1А).
Рекомендация 4.3. Клинико-патологическая диагностика различных вариантов МБПГН должна включать определение общего уровня сывороточного комплемента (СН50), а также его фракций в сыворотке крови: С3 и С4 (1А).
Рекомендация 4.4. Нормальный уровень С4-фракции комплемента свидетельствует об альтернативном пути активации комплемента (иммуноглобулиннегативный, С3-позитивный МБПГН), а снижение его концентрации - о классическом пути активации комплемента (иммуноглобулинпозитивный, С3-позитивный МБПГН). В обоих указанных случаях снижен общий уровень сывороточного комплемента (СН50) и его С3-фракции (1А).
Рекомендация 4.5. Для более полного суждения о патогенезе иммуноглобулиннегативного, С3-позитивнго МБПГН I или III типов и БПД необходимо определять в сыворотке крови титр С3-нефритического фактора, исследовать уровень регуляторных протеинов альтернативного пути активации комплемента: факторы Н, I, В, пропердин (1А).
Рекомендация 4.6. Иммуноглобулин- и С3-негативный вариант МБПГН следует рассматривать как репаративную фазу воспалительного процесса, обусловленного первичным повреждением эндотелиоцитов (2В).
Рекомендация 4.7. При иммуноглобулин- и С3-негативном варианте МБПГН концентрация общего уровня комплемента в сыворотке крови (СН50) и его фракций (С3, С4) не меняется (1А).
Комментарий. Иммуноглобулин- и комплементпозитивный вариант МБПГН I и III типов (см. рис. 1), как правило, носят вторичный характер и связаны с хронической антигенемией, циркуляцией в крови аутоиммунных комплексов или с отложением в гломеруле моноклональных иммуноглобулинов. В сравнительно редких случаях, когда не удается установить причину хронической антигенемии, подтвердить наличие плазмаклеточной дискразии или аутоиммунного процесса, допускается диагностика идиопатической формы МБПГН I или III типа. Причиной хронической антигенемии, как правило, являются торпидно протекающие вирусные, бактериальные, протозойные и прочие инфекции (табл. 1).
Патогенез иммуноглобулинпозитивного МБПГН I и III типов имеет общие черты. Иммунные комплексы, образовавшиеся в циркуляции крови или in situ, вследствие хронической антигенемии (инфекции), или циркулирующие ИК при аутоиммунных процессах [СКВ, синдром Съегрена, смешанная криоглобулинемия (КГ) и др.], или ИК, сформировавшиеся при парапротеинемиях (моноклональные гаммапатии, лимфопролиферативные заболевания), откладываются в гломерулах мезангиально (при крупных размерах), субэндотелиально (при средних размерах) или субэпителиально (при мелких размерах).
|
|
Иммунные комплексы активируют комплемент по классическому пути, который задействует фракции комплемента Clq, C2, C4 с образованием С3-конвертазы классического пути (С4bС2а), которая расщепляет С3-фракцию на С3а- и С3b-субфракции с последующим образованием С5-конвертазы классического пути активации комплемента (С4bС2аС3b). С5-конвертаза, действуя на С5-фракцию комплемента, приводит к образованию С5а- и С5в-подфракций, причем последняя в конечном итоге приводит к образованию мембрано-атакующего комплекса (МАК) (С5b-9). Субфракции комплемента С3а и С5а, действуя хемотаксически, обусловливают приток к месту расположения ИК макрофагов и нейтрофилов из циркулирующей крови, которые за счет провоспалительных цитокинов и протеолитических энзимов обусловливают формирование в гломеруле экссудативно-воспалительной реакции. Резидентные клетки клубочка (эндотелиоциты, мезангиоциты) на повреждение провоспалительными цитокинами и цитопатическое действие МАК (С5b-9) отвечают пролиферацией, синтезом основного вещества (базальные мембраны, мезангиальный матрикс) и продукцией ростовых факторов (трансформирующий фактор роста β1, тромбоцитарный фактор роста). В конечном итоге формируются морфологические признаки в виде удвоения базальных мембран, пролиферации мезангиоцитов и мезангиального матрикса с лобулизацией клубочка, образования зон склероза (клубочки и тубулоинтерстиций). Заметим, что вторичный МБПГН при HCV-инфекции может иметь двоякий патогенез. В одних случаях он может быть связан с образованием ИК к антигенам HCV, отложившихся первоначально в клубочке (т.е образующихся in situ), в других случаях речь идет о циркулирующих ИК - смешанных криоглобулинах (II тип криоглобулинемии). Смешанные криоглобулины (II типа) при HCV-инфекции - это иммунные комплексы, преципитирующие на холоде, состоящие из IgMK (ревматоидный фактор). Наличие смешанной криоглобулинемии, ассоциированной с HCV-инфекцией, рассматривается некоторыми авторами как субклиническая форма лимфомы. Среди иммуноглобулинпозитивных вариантов МБПГН особое место занимает трансплантационная гломерулопатия. Долгое время патоморфологические изменения в трансплантированной почке рассматривали с точки зрения механизмов хронического отторжения трансплантата (хроническая трансплантационная нефропатия). В настоящее время накоплены научные данные, позволяющие выделить трансплантационную гломерулопатию в самостоятельную клинико-морфологическую нозологическую единицу с иммунным патогенезом. Трансплантационная гломерулопатия представляет собой первоначальное повреждение эндотелиоцитов аутоантителами к человеческим лейкоцитарным антигенам (HLA) II класса, которые присутствуют на наружной клеточной мембране эндотелиальных клеток. В острой фазе развивается так называемый гломерулит, характеризующийся повреждением гломерулярных капилляров, мигрирующими из циркулирующей крови мононуклеарами и нейтрофилами. Острую экссудативную реакцию в клубочке (гломерулит) сменяет репаративная фаза, в которую происходит пролиферация и экспансия мезангиального матрикса, развивается дупликация базальных мембран, и морфологическая картина при световой микроскопии становится аналогичной иммуноглобулинпозитивному МБПГН. При иммунофлюоресценции регистрируется отложение вдоль петель капилляров клубочка фракции комплемента С4d - продукта активации комплемента по классическому пути, однако даже отсутствие депозитов С4d не будет противоречить диагнозу трансплантационной гломерулопатии.
Этиология иммуноглобулин-негативного, С3-позитивного ГН, называемого С3-гломерулопатией, объясняется дисрегуляцией альтернативного пути активации комплемента и нарушением терминальной стадии образования МАК (С5b-9). Нарушение нормальной физиологии альтернативного пути активации комплемента может быть обусловлено либо мутацией генов различных факторов системы комплемента, либо носить приобретенный характер. В последнем случае в организме формируются аутоантитела к регуляторным факторам активации комплемента по альтернативному пути. Химическая структура депозитов при С3-гломерулопатии окончательно не установлена, но выяснено, что они состоят из гликозоаминогликанов с включениями С3b-фракции комплемента, продуктов ее деградации (iC3b, C3dg, C3c), а также компонентов МАК (С5b-9). В отличие от классического пути активации комплемента, когда реакции каскадного типа запускаются ИК, для альтернативного пути и в норме свойственна постоянная, персистирующая активность низкой степени, заключающаяся в образовании небольших количеств С3b-фракции, вследствие спонтанного гидролиза тиоэфирной связи С3-протеина. Генерируемая в небольших количествах фракция С3b-комплемента далее связывается с мембранами различных клеток, в том числе с мембранами патогенных микроорганизмов, в чем состоит физиологический смысл данной реакции. С целью предотвращения перехода данной спонтанной активности в неуправляемую реакцию (каскад), в организме существует целая система регуляторных факторов (протеинов), действующих на различных уровнях каскадной реакции, особенно при образовании С3- и С5-конвертаз. Фактор «Н» (CFH) способствует распаду спонтанно образующейся С3-конвертазы альтернативного пути (C3bВb) и совместно с фактором «I» (CFI) (для которого CFH является кофактором) приводит к инактивации субфракции С3b. В регуляции системы активации комплемента по альтернативному пути в циркулирующей крови (регуляторы «жидкой фазы») принимает участие также группа протеинов (от 1 до 5), подобных фактору «Н» (CFHR 1-5 - complement factor H related proteins). Функция их окончательно не изучена. Полагают, что CFHR1 ингибирует действие МАК, а механизм действия CFHR5 аналогичен регуляторной активности фактора «Н». Причиной формирования С3-позитивного МБПГН, в том числе БПД, могут являться мутации гена фактора «Н». Моногенная мутация CFHR5, наследуемая по аутосомно-доминантному пути, является причиной эндемической Кипрской нефропатии, представляющей собой С3-позитивный МБПГН I или III типа.
Необходимо отметить, что факторы «Н» и CFHR5, действуя в плазме крови, обладают также тропностью к экстрацеллюлярным мембранам, где сохраняют свою инактивирующую активность по отношению к мембраносвязанной субфракции комплемента С3b. Из данного факта вытекает несколько важных для понимания патогенеза С3-позитивной гломерулопатии обстоятельств. Известно, что патогенез атипичного гемолитико-уремического синдрома (аГУС) также может быть связан с генетическими мутациями регуляторного фактора «Н». Однако при этом заболевании дисрегуляция альтернативного пути активации комплемента происходит главным образом на поверхности клеточных мембран эндотелиоцитов, не затрагивая систему активации комплемента в циркулирующей крови. Поэтому, хотя в редких случаях и возможно первоначальное формирование С3-позитивной гломерулопатии при аГУС, наиболее типичным сценарием патологического при нем процесса является инициальное повреждение эндотелиоцитов с формированием микротромбозов капилляров клубочков и лишь спустя некоторое время, когда активируются репаративные (пролиферативные) процессы как ответная реакция резидентных клеток клубочка на эндотелиальное повреждение, начинает формироваться морфологическая картина МБПГН (С3-негативного и без отложений электронноплотных депозитов). CFHR5 обладает сродством к гликозоаминогликанам, а поэтому при мутации гена этого фактора (Кипрская нефропатия) происходит первичная активация альтернативного пути комплемента на гломерулярной базальной мембране. В результате формируется С3-позитивный МБПГН с субэндотелиальными и/или субэпителиальными электронноплотными депозитами (I или III типа). Ингибирующее действие факторов «Н» и CFHR5 в отношении C3b на поверхности гломерулярной базальной мембраны формирует физиологическую защиту почек от иммунокомплексного ГН и объясняет те редкие случаи иммуноглобулинпозитивного МБПГН (т.е. иммунокомплексного), при котором выявляют мутации гена фактора «Н». В литературе описаны также мутации генов основных белков системы комплемента. Так, при гетерозиготной мутации С3-протеина, в плазме крови присутствуют как мутантный С3-белок, так и нативный, синтезируемый геном аллели, не вовлеченной в мутацию. В результате спонтанного гидролиза мутантного С3-протеина образуется С3-конвертаза, резистентная к действию фактора «Н», которая расщепляет С3-белок, синтезируемый нормальным геном, вследствие чего образуются в избытке продукты деградации С3-фракции комплемента, что запускает каскадную реакцию активации комплемента по альтернативному пути. Подобный механизм может лежать в основе ответной гломерулярной реакции в виде формирования БПД. Генетический полиморфизм факторов системы комплемента, ведущий к изменению структуры протеинов и к нарушению их функции, также может играть не последнюю роль в патогенезе С3-позитивной гломерулопатии.
Следует подчеркнуть, что система комплемента имеет многоступенчатую систему регуляции, а поэтому не всякая генетическая мутация или генный полиморфизм реализуется клинически. В большинстве случаев необходимо сочетанное действие факторов внешней среды для формирования генетически запрограммированного фенотипа. К числу таких провоцирующих факторов прежде всего следует отнести инфекции, а возможно, и другие причины (образ жизни, питания, хронические интоксикации, сопутствующие заболевания и др.). Подтверждением сказанному могут служить хорошо известные клиницисту случаи синфарингитической макрогематурии при МБПГН.
Причина приобретенных нарушений в системе регуляции альтернативного пути активации комплемента заключается в образовании в организме аутоантител к регуляторным протеинам (факторы Н, В и др.) или к основным фракциям комплемента. Наиболее известным и изученным является С3-нефритический фактор (C3NeF), представляющий собой аутоантитело (IgG) к С3-конвертазе (C3bBb) альтернативного пути активации комплемента. Присоединение аутоантитела к С3-конвертазе делает ее более устойчивой к действию регуляторных протеинов (CFH, фактор I, CFHR 1-5), что продлевает время ее циркуляции в крови.
Итогом нерегулируемой деятельности С3-конвертазы является активация комплемента с постепенным истощением пула С3-фракции и снижением ее концентрации в плазме крови. С3NeF обнаруживается у 86% пациентов с БПД и у 49% больных с С3-позитивным ГН, однако не у всех больных это сочетается со снижением С3-фракции комплемента, что говорит о существовании других регуляторных механизмов в организме, противодействующих С3NeF. С наличием дисрегуляции альтернативного пути активации комплемента при БПД связывают два состояния, часто ассоциирующихся с этим заболеванием. Первое представлено приобретенной частичной липодистрофией (acquired partial lipodystrophy), клинически характеризующейся постепенной (в течение многих лет) симметричной потерей подкожно-жировой клетчатки в цефалокаудальном направлении, начиная с лица, шеи, рук, грудной клетки. На завершающей стадии может вовлекаться подкожно-жировая клетчатка нижних конечностей. Полагают, что С3NeF вызывает активацию комплемента на клеточной поверхности адипоцитов, что приводит к их гибели через апоптоз. Второе состояние характеризуется образованием беловато-желтых друз (бляшек) в пигментной оболочке сетчатки глаза. Визуальная картина глазного дна и клиническое течение аналогично возрастной макулярной дистрофии сетчатки. Считается, что ведущим патогенетическим механизмом данного процесса является нарушение местной регуляторной активности фактора «Н». При электронной микроскопии аутопсийного материала (сетчатки глаза) выявляются электронноплотные депозиты вдоль базальных мембран капилляров сетчатой оболочки. Вследствие хориоидальной неоваскуляризации, развивающейся со временем, отмечается постепенная потеря зрения.
Причина того факта, что в одном случае С3-позитивной гломерулопатии формируется морфологическая картина МБПГН I или III типа, а в другом - выявляется БПД, остается невыясненной. По-видимому, имеют значение гетерогенность генетических мутаций, первоначальная локализация процесса, степень активации системы комплемента. Активация альтернативного пути комплемента, как говорилось выше, может быть задействована и в случаях первичного иммунокомплексного механизма повреждения, особенно в том случае, когда основному патологическому процессу сопутствует генетический полиморфизм генов регуляторных протеинов (CFH, CFI). При моноклональных гаммапатиях, при которых обычно формируется иммуноглобулинпозитивный МБПГН (для которого характерен классический путь активации комплемента), недавно был открыт иной путь патогенеза. Оказалось, что моноклональный иммуноглобулин может действовать как АТ к фактору «Н» и к другим регуляторным протеинам, приводя к дисрегуляции альтернативного пути комплемента и к формированию С3-позитивной гломерулопатии.
Этиология иммуноглобулин- и С3-негативного МБПГН заключается в первичном поражении эндотелиоцитов (ТМА, синдром злокачественной гипертензии и др.), за которым следует репаративная фаза в форме пролиферативных изменений в клубочке, идентифицируемых светооптически, как МБПГН. При электронной микроскопии в этих случаях не выявляются электронноплотные депозиты, а следовательно, установить тип МБПГН не представляется возможным (см. рис. 1).
Причины иммуноглобулин- и С3-комплементнегативного мембранопролиферативного гломерулонефрита:
Морфопатогенез С3-негативной гломерулопатии при большинстве заболеваний сводится к повреждению эндотелиоцитов в острой фазе, что проявляется их набуханием, развивается мезангиолизис, образуются фибриновые тромбы в капиллярах клубочках. Острую фазу повреждения сменяет репаративная фаза, характеризующаяся ответной реакцией резидентных клеток клубочка. Происходит увеличение мезангиального матрикса и пролиферация мезангиальных клеток, появляются двухконтурные базальные мембраны капилляров, т.е. формируется морфологическая картина МБПГН.
В редких случаях генетической аномалии - дефицита а1-антитрипсина - в печени синтезируется мутантный протеин Z, который, попадая в гломерулы с циркулирующей кровью, полимеризируется и откладывается субэндотелиально. Депозиты Z-протеина являются причиной ответной реакции резидентных клеток клубочка, которая на завершающем этапе приводит к формированию морфологической картины МБПГН при световой микроскопии. Уточнить диагноз можно при иммунофлюоресценции с использованием специфических антисывороток к Z-протеину.
5. Лечение идиопатического мембранопролиферативного гломерулонефрита
Рекомендация 5.1. При решении вопроса о характере патогенетической терапии идиопатического МБПГН необходимо принимать во внимание ведущий клинический синдром и данные морфологического исследования биоптатов почки (НГ).
Рекомендация 5.2. Иммуносупрессивная терапия при идиопатическом МБПГН показана только в случаях с НС, при медленно прогрессирующем, но неуклонном падении функции почек, несмотря на проводимую нефропротективную терапию, или при быстропрогрессирующем нефритическом синдроме (2D).
Рекомендация 5.3. Наиболее оптимальной схемой иммуносупрессивной терапии идиопатического МБПГН при НС или при медленно прогрессирующем падении функции почек является применение циклофосфамида (2-2,5 мг/кг/сут) или микофенолата мофетила (1,5-2 г/сут) в сочетании с преднизолоном (40 мг/сут) по альтернирующей схеме. Продолжительность терапии должна составлять не менее 6 мес (2D).
Рекомендация 5.4. При идиопатическом МБПГН с быстропрогрессирующим нефритическим синдромом показан плазмаферез (по 3 л плазмы за сеанс 3 раза в неделю), пульс-терапия метилпреднизолоном (0,5-1,0 г/сут 3 дня) и далее поддерживающая иммуносупрессивная терапия по схеме (см. рекомендацию 5.3) (2D).
Комментарий. В отношении тактики лечения иммуноглобулинпозитивного идиопатического МБПГН в настоящее время нет единой точки зрения. При решении вопроса о характере патогенетической терапии идиопатического МБПГН необходимо принимать во внимание клинический вариант течения болезни (ведущий клинический синдром) и данные морфологического исследования биоптатов почки. Если в клинической картине доминирует изолированный мочевой синдром или синдром рецидивирующей макрогематурии, ограничиваются ренопротективной терапией (ИАПФ, АТ1 - антагонисты, статины, диета) и стремятся к полной нормализации АД (не выше 130/80 мм рт.ст.). Если у больного отмечается субнефротическая ПУ (менее 3,5 г/сут) и снижение функции почек до уровня ХБП 3-4-й стадии, а при морфологическом исследовании выявляется выраженный тубулоинтерстициальный склероз, дополнительно могут быть назначены аспирин (975 мг/день) и дипиридамол (325 мг/день) (доказательная база эффективности такой терапии отсутствует). В случаях НС и прогрессирующего ухудшения функции почек применяют комбинацию циклофосфамида (2-2,5 мг/кг в сутки) или микофенолата мофетила (1,5-2 г/сут) в сочетании с низкими дозами преднизолона (40 мг/сут) лучше по альтернирующей схеме в течение 6 мес (рекомендации KDIGO). При БПНС с наличием полулуний более чем в 50% клубочков рекомендуется плазмаферез, пульс-терапия метилпреднизолоном с последующим пероральным приемом циклофосфамида в сочетании с преднизолоном (схему см. выше). Подчеркнем, что при всех клинических вариантах течения МБПГН всегда проводятся мероприятия по ренопротекции.
6. Лечение вторичного мембранопролиферативного гломерулонефрита
Рекомендация 6.1. При вторичных формах МБПГН основным направлением в лечении является терапия основного заболевания (см. табл. 1) (1А).
Рекомендация 6.2. Применение иммуносупрессии при вторичных формах МБПГН допускается только в случаях с быстропрогрессирующим нефритическим синдромом (2В).
Комментарий. При иммуноглобулин-позитивном МБПГН прежде всего необходимо установить или исключить вторичную причину заболевания (см. табл. 1). При вторичных формах МБПГН главным условием остается лечение основного заболевания. Особенно это касается инфекций. При HCV-ассоциированном МБПГН с ХБП 1-й и 2-й стадии вне зависимости от патогенеза (некриоглобулинемический или криоглобулинемический варианты) первой линией терапии является применение пегилированных интерферонов-α и рибавирина в обычных дозах с учетом генотипа вируса. При ХБП 3, 4 и 5-й стадии (вне зависимости от диализной терапии) рекомендуется: пэгинтерферон альфа 2a: 135 мкг подкожно 1 раз в неделю или пэгинтерферон альфа 2b: 1 мкг/кг подкожно 1 раз в неделю. Согласно последним рекомендациям KDIGO, рибавирин следует использовать с осторожностью при СКФ <50 мл/ мин/1,73 м2 (табл. 2).
При криоглобулинемическом варианте МБПГН, который резистентен к применению антивирусных препаратов или протекает с выраженными признаками криоглобулинемического васкулита (кожа, легкие, ГН с полулуниями), препаратом выбора является ритуксимаб (анти-CD-20 моноклональное АТ), применение которого приводит к истощению пула В-лимфоцитов, продуцирующих криоглобулины (375 мг/м2 1 раз в неделю в течение 4 нед).
| Стадия ХБП | Интерферон | Рибавиринб |
|---|---|---|
1 и 2 |
Пэгинтерферон альфа 2a: 180 мкг п/к еженедельно. Пэгинтерферон альфа 2b: 1,5 мкг/кг п/к еженедельно |
800-1200 мг/день, разделенные на 2 дозы |
3 и 4 |
Пэгинтерферон альфа 2a: 135 мкг п/к еженедельно. Пэгинтерферон альфа 2b: 1 мкг/кг п/к еженедельно* |
|
5 |
Пэгинтерферон альфа 2a: 135 мкг п/к еженедельно. Пэгинтерферон альфа 2b: 1 мкг/кг п/к еженедельно* |
Примечание: п/к - подкожно.
а - Пациенты с генотипами 1 и 4 должны получать терапию интерферонами в течение 48 нед, если ранний вирусный/вирусологический ответ достигнут в течение 12 нед (снижение титра вируса >2 log). Генотипы 2 и 3 должны получать терапию в течение 24 нед.
б - Пациенты с генотипами 2 и 3 должны получать 800 мг/день на стадиях 1 и 2 ХБП. Инфицированные пациенты с генотипами 1 и 4 должны получать 1000-1200 мг/день на стадиях 1 и 2 ХБП
* Со времени публикации руководства KDIGO по гепатиту С у больных ХБП инструкция по применению лекарственного препарата изменилась, и теперь разрешается одновременное/совместное применение рибавирина у пациентов с ХБП 3-5-й стадий, если побочные эффекты выражены минимально и поддаются коррекции. При клиренсе креатинина <50 мл/мин рекомендуется соблюдать осторожность, что может потребовать существенного снижения дозы. Информация о модификации дозы изложена в инструкции по применению препарата.
Менее эффективной альтернативой в этих случаях является плазмаферез (3 л плазмы 3 раза в неделю, 2-3 нед) в сочетании с пульс-терапией метилпреднизолоном (0,5-1 г/сут 3 дня), преднизолоном (1-1,5 мг/кг в день) и циклофосфамидом (2 мг/кг в день) в течение 2-4 мес. Дозы препаратов следует соотносить со значениями СКФ. При некриоглобулинемическом HCV-ассоциированном МБПГН от иммуносупрессии следует воздержаться, за исключением случаев с БПНС и наличием полулуний в клубочках. При бактериальных инфекциях (например, при ИЭ) иммуносупрессия не рекомендуется (рекомендации KDIGO). При остальных заболеваниях, перечисленных в табл. 1 и являющихся причиной вторичного МБПГН, проводят лечение основной болезни.
При иммуноглобулиннегативных вариантах МБПГН лечение назначается с учетом данных о патогенезе заболевания. При С3-позитивной гломерулопатии, обусловленной мутациями генов регуляторных факторов системы комплемента (H, I), показаны инфузии свежезамороженной донорской плазмы крови (донатор нативных факторов). Если причиной С3-позитивной гломерулопатии являются аутоантитела к С3-конвертазе (С3NeF), регуляторным факторам H, I и др., лечение целесообразно начинать с плазмафереза (в режиме плазмаобмена и с использованием замещающего раствора в виде донорской плазмы и альбумина). Далее, как правило, показаны ГК или ритуксимаб (блокируют выработку аутоантител). В последнее время появились работы о высокой эффективности при генетических вариантах С3-позитивной гломерулопатии экулизумаба, представляющего собой моноклональное АТ к С5-фракции комплемента (блокирует образование МАК). Как известно, экулизумаб изначально был предложен для лечения пароксизмальной ночной гемоглобинурии и аГУС. При других патогенетических вариантах С3-негативной гломерулопатии тактика терапии зависит и определяется основным заболеванием.
7. Прогноз
Рекомендация 7.1. При определении прогноза МБПГН необходимо принимать во внимание клинические, лабораторные и морфологические факторы (см. Предикторы неблагоприятного прогноза в отношении почечной выживаемости при иммуноглобулинпозитивном мембранопролиферативном гломерулонефрите) (2С).
Комментарий. Определить точно прогноз в отношении развития МБПГН затруднительно, поскольку за последние годы изменились представления о патогенезе болезни, что не позволяет использовать исторический контроль. 10-летняя почечная выживаемость для иммуноглобулинпозитивного МБПГН, по-видимому, составляет 50-60% и зависит от многих факторов (см. Предикторы неблагоприятного прогноза…), и главным из них является формирование полулуний более чем в 50% клубочков.
При С3-гломерулопатии 10-летняя почечная выживаемость составляет 30-50% (при генетических вариантах - более низкая). Частота возвратного ГН в трансплантате при иммуноглобулинпозитивном МБПГН колеблется в пределах 18-50% (прогностически неблагоприятным предиктором является HLA гаплотип B8DR3). Выживаемость трансплантата может быть улучшена путем добавления к иммуносупрессивной терапии циклофосфамида. При БПД частота возвратного ГН составляет от 67 до 100%.
Если причиной БПД является мутация гена фактора «Н», показаны плазмаферез и инфузии свежезамороженной плазмы (СЗП) до и после операции ТП.
Предикторы неблагоприятного прогноза в отношении почечной выживаемости при иммуноглобулинпозитивном мембранопролиферативном гломерулонефрите
Клинические:
Лабораторные:
Морфологические:
СПИСОК ЛИТЕРАТУРЫ
-
Добронравов В.А., Дунаева Н.В. Поражение почек и хронический вирусный гепатит С // Нефрология. 2008. Т. 12, № 4. С. 9-19.
-
Лора Ш., Фремю-Бачи В. Атипичный гемолитико-уремический синдром // Нефрология. 2012. Т. 16, № 2. С. 16-48.
-
Ферри С. Смешанная криоглобулинемия // Нефрология. 2010. Т. 14, № 1. С. 11-28.
-
Appel G.B. Membranoproliferative glomerulonephritis - mechanisms and treatment // Contrib. Nephrol. 2013. Vol. 181. P. 163-174.
-
Bomback A.S., Appel G.B. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN // Nat. Rev. Nephrol. 2012. Vol. 8. P. 634-642.
-
Bomback A.S., Smith R.J., Barile G.R. et al. Eculisumab for dense deposit disease and C3 glomerulonephritis // Clin. J. Am. Soc. Nephrol. 2012. Vol. 7. P. 748-756.
-
D’Agati V.D., Bomback A.S. C3 glomerulopathy: what’s in a name? // Kidney Int. 2012. Vol. 82. P. 379-381.
-
Fervensa F.C., Sethi S., Glassock R.J. Idiopathic membraneproliferative glomerulonephritis: does it exist? // Nephrol. Dial. Transpant. 2012. Vol. 27, N 12. P. 4288-4294.
-
Fregonese L., Stolk J. Hereditary alpha-1-antitrypsin deficiency and its clinical conseguence // Orphanet J. Rare Dis. 2008. Vol. 3. P. 16.
-
Hou J., Markowitz G.S., Bomback A.S. et al. Toward a working definition of C3 glomerulopathy by immunofluorescence // Kidney Int. 2013. Sept 25. [Epub ahead of print].
-
KDIGO Clinical practice guideline for glomerulonephritis // Kidney Int. Suppl. 2012. Vol. 2, N 2. P. 198-200.
-
Morales J.M., Kamar N., Rostaing L. Hepatitis C and renal disease: epidemiology, diagnosis, pathogenesis and therapy // Contrib. Nephrol. 2012. Vol. 176. P. 10-23.
-
Pickering M.C., Cook H.H. Complement and glomerular disease: new insights // Curr. Opin. Nephrol. Hypertens. 2011. Vol. 20. P. 271-277.
-
Pickering M.C., D’Agati V.D., Nester C.M. et al. C3 glomerulopathy: consensus report // Kidney Int. 2013. Oct 30 [Epub ahead of print].
-
Servias A., Noel L-H., Roumenina L.T. et al. Acguired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glo-merulopathies // Kidney Int. 2012. Vol. 82. P. 454-464.
-
Sethi S., Fervenza F.C. Memranoproliferative glomerulonephritis - a new look at an old entity // N. Engl. J. Med. 2012. Vol. 366. P. 1119-1131.
-
Smith R.J.H., Harris C.Z., Pickering M.C. Dense deposit disease // Mol. Immunol. 2011. Vol. 48. P. 1604-1610.
-
Sun Q., Huang X., Jiang S. et al. Picking transplant glomerulopathy out of the CAN: evidence from a clinico-pathological evaluation // BMC Nephrol. 2012. Vol. 13. P. 128.
МЕЗАНГИОПРОЛИФЕРАТИВНЫЙ ГЛОМЕРУЛОНЕФРИТ (IgA-НЕФРОПАТИЯ)
КОДИРОВАНИЕ ПО МЕЖДУНАРОДНОЙ КЛАССИФИКАЦИИ БОЛЕЗНЕЙ 10-ГО ПЕРЕСМОТРА
Класс XIV: Болезни мочеполовой системы.
Блок N00-N08: Гломерулярные болезни.
N00.3. Острый нефритический синдром - диффузный мезангиальный пролиферативный ГН.
N01.3. Быстропрогрессирующий нефритический синдром - диффузный мезангиальный пролиферативный ГН.
N02.3. Рецидивирующая и устойчивая гематурия - диффузный мезангиальный пролиферативный ГН.
N03.3. Хронический нефритический синдром - диффузный мезангиальный пролиферативный ГН.
N04.3. НС - диффузный мезангиальный пролиферативный ГН
N05.3. Нефритический синдром неуточненный - диффузный мезангиальный пролиферативный ГН.
N06.3. Изолированная ПУ с уточненным морфологическим поражением - диффузный мезангиальный пролиферативный ГН.
N07.3. Наследственная нефропатия, не классифицированная в других рубриках - диффузный мезангиальный пролиферативный ГН
1. Введение
IgA-нефропатия (синонимы - IgA-нефрит, болезнь Берже, синфарингитная гематурия) - иммунокомплексный гломерулонефрит (ГН), характеризующийся преимущественным отложением в мезангии иммуноглобулина А (IgA), при классическом течении - мезангиальной пролиферацией.
ЭПИДЕМИОЛОГИЯ
Представляет собой наиболее распространенную форму первичного (идиопатического) ГН в мире: по данным морфологических регистров, частота IgA-нефропатии варьирует в зависимости от географического региона, составляя от 10-20% в США и Европе, до 40-45% - в странах Азии [47, 53, 60]. По-видимому, истинная распространенность IgA-нефропатии выше, так как, с одной стороны, не всем пациентам с характерной клинической картиной заболевания выполняют биопсию почки, с другой - признаки IgA-нефропатии обнаруживают в биоптатах почек у лиц без явных клинических проявлений заболевания [69, 75]. IgA-нефропатия чаще развивается у представителей азиатской и европеоидной расы [35]. Она может начинаться в любом возрасте, однако пик заболеваемости приходится на второе и третье десятилетия жизни [15, 33]. Мужчины заболевают в среднем в 2 раза чаще, чем женщины. IgA-нефропатия в 35-50% случаев рецидивирует в трансплантате; выживаемость трансплантата выше, чем при других болезнях почек [11, 58].
ПАТОГЕНЕЗ IgA-НЕФРОПАТИИ
Механизм развития IgA-нефропатии - иммунокомплексный. По современным представлениям, ведущую роль играют изменения структуры молекулы IgA, обусловленные нарушением процессов ее гликозилирования и полимеризации и, как следствие, нарушение взаимодействия с белками матрикса, рецепторами к IgA на мезангиальных и других клетках, с компонентами системы комплемента. Это приводит к депонированию полимерного IgAl в мезангии, активации синтеза клетками почек различных цитокинов и факторов роста с развитием характерных морфологических изменений [7, 61, 68].
Предрасположенность к IgA-нефропатии может быть обусловлена носительством определенных локусов системы HLA, в частности, для семейных форм идентифицированы локусы 6q22-23 (IGAN1), 4q26-31 (IGAN2) и 17q12-22 (IGAN3) [5, 28]. Уточняется роль других возможных генов-кандидатов.
Провоцирующими факторами являются инфекции верхних и нижних дыхательных путей, острые инфекционные или вирусные гастроэнтериты, другие инфекции, вакцинация, ультрафиолетовое облучение.
КЛИНИЧЕСКАЯ КАРТИНА
Клиническая картина IgA-нефропатии разнообразна: проявления варьируют от изолированной бессимптомной микрогематурии до быстропрогрессирующего ГН [18-21].
Основные клинические проявления IgA-нефропатии.
-
Гематурия - разной степени выраженности (микрогематурия, в 40-50% случаев макрогематурия), может быть изолированной и в сочетании с протеинурией (ПУ), при фазово-контрастной микроскопии осадка мочи выявляются дисморфные эритроциты.
-
ПУ - обычно небольшая (<1 г/сут), изолированной бывает редко. Возможно развитие выраженной ПУ с формированием нефротического синдрома (НС) на разных стадиях болезни. НС выявляется только у 5% всех больных IgA-нефропатией, более характерен для детей и подростков. Выраженность ПУ определяет прогноз заболевания.
-
АГ - чаще наблюдается при высокоактивном течении ГН в сочетании с ПУ и острой почечной недостаточностью (ОПН) (в дебюте или при обострении нефрита), также развивается при формировании нефросклероза.
-
Острая ПН (в сочетании с олигурией, отеками и АГ) может возникнуть на любой стадии IgA-нефропатии в результате тяжелого иммунного повреждения клубочков с формированием полулуний или вследствие окклюзии канальцев эритроцитами.
-
Хроническая ПН развивается при длительном течении заболевания, в основном, у пациентов с высоким риском прогрессирования хронического гломерулонефрита (ХГН).
-
Повышение уровня IgA (в основном полимерных форм) в сыворотке крови обнаруживается у 35-60% больных, как правило, не коррелирует с тяжестью болезни.
Различают следующие варианты клинического течения IgA-нефропатии.
-
Классический с повторными эпизодами макрогематурии (синфарингитная гематурия) (30-50% больных):
-
характерно острое начало с эпизода макрогематурии, возникает на фоне инфекционного заболевания (обычно респираторной, реже мочевой, кишечной инфекции и т.д.) одновременно или в первые 2-3 дня болезни и сохраняется от нескольких часов до нескольких дней;
-
при микроскопическом исследовании осадка мочи обнаруживают дисморфные эритроциты, эритроцитарные цилиндры;
-
в промежутках между эпизодами макрогематурии сохраняется персистирующая микрогематурия или наблюдается полная нормализация анализов мочи (до следующего эпизода).
-
Атипичные формы протекают с клинической картиной, в большей степени сходной с другими вариантами гломерулярного повреждения - БМИ с IgA-депозитами в мезангии; острое повреждение почек (ОПП), ассоциированное с эпизодами макрогематурии; быстропрогрессирующий ГН с полулуниями и депозитами IgA в мезангии.
Рекомендация 1. Диагностика IgA-нефропатии базируется на выявлении специфических изменений при морфологическом исследовании ткани почки (НГ).
МОРФОЛОГИЧЕСКИЕ КРИТЕРИИ IgA-НЕФРОПАТИИ
При световой микроскопии спектр морфологических изменений широкий и варьирует, в том числе, в биоптатах отдельного пациента [29]:
-
характерны очаговая или диффузная мезангиальная пролиферация с расширением внеклеточного матрикса;
-
возможны различной степени выраженности интра- или экстракапиллярная пролиферация;
-
возможны сегментарный некроз капилляров клубочка и образование полулуний;
-
на поздних стадиях болезни отмечается интерстициальный фиброз, ангиосклероз, атрофия канальцев.
Иммунофлюоресцентное исследование - основа диагностики IgA-нефропатии. Выявляют депозиты IgA (часто в сочетании с депозитами и/или IgG) в мезангии и в капиллярных стенках клубочков. По данным различных центров, частота выявления изолированных депозитов IgA колеблется от 0 до 85%. Почти всегда обнаруживают депозиты С3, реже С4; наличие депозитов Clq не характерно.
При электронной микроскопии характерны отложения электронно-плотного материала в мезангии, реже субэндотелиально или субэпителиально, соответствующие иммунным комплексам, определяемым при иммунофлуоресцентной микроскопии.
2. Дифференциальная диагностика
Рекомендация 2. Диагноз первичной IgA-нефропатии должен базироваться на исключении вторичного характера заболевания (НГ).
IgA-нефропатию можно заподозрить на основании характерной клинической картины: наличие эпизодов синфарингитной гематурии и/или персистирующей микрогематурии в сочетании с ПУ различной степени выраженности и повышенным уровнем IgA в крови.
Необходим тщательный сбор анамнеза, комплексная оценка результатов клинического, лабораторного и инструментального обследования для исключения вторичных причин IgA-нефропатии. При вторичной форме IgA-нефропатии на момент биопсии, как правило, уже имеются клинические и лабораторные признаки основного заболевания.
Причины вторичной IgA-нефропатии
-
Заболевания кишечника: целиакия, болезнь Крона, неспецифический язвенный колит.
-
Заболевания легких и бронхов: саркоидоз, муковисцидоз, идиопатический гемосидероз легких, облитерирующий бронхиолит.
-
Инфекции и паразитарные заболевания: ВИЧ-инфекция, HBV-инфекция, HCV-инфекция, диссеминированный туберкулез, лепра, хронический шистосомоз, токсоплазмоз
-
Другие системные и аутоиммунные заболевания: геморрагический васкулит Шенлейна-Геноха, СКВ, ревматоидный артрит, анкилозирующий спондилит, КГ, склеродермия, синдром Шегрена, болезнь Бехчета, синдром Рейтера
-
Заболевания, которые могут сочетаться с IgA-нефропатией: АНЦА-васкулиты (гранулематоз Вегенера), диабетическая нефропатия (ДН), мембранозная нефропатия (МН).
IgA-нефропатию следует дифференцировать с другими мезангиальными формами ГН и рядом наследственных нефропатий, протекающих с гематурией [синдром Альпорта (СА), болезнь тонких мембран].
Клинические проявления IgA-нефропатии не различаются при идиопатическом и вторичном вариантах заболевания, в этой связи дифференциальная диагностика этих форм должна базироваться на исключении всех возможных вторичных причин заболевания.
Необходимо провести полноценное урологическое обследование для исключения опухолей почек и мочевыводящих путей, аномалий строения и положения почек, мочекаменной болезни, которые могут симулировать клиническую картину IgA-нефропатии, а также сосуществовать с ней.
3. Лечение
Специфического (этиологического) лечения первичной IgA-нефропатии в настоящее время не существует, основная цель терапии - отсрочить начало заместительной поченой терапии (ЗПТ) [39].
Рекомендация 3. Тактика лечения больных IgA-нефропатией основывается на оценке риска прогрессирования с помощью клинико-лабораторных и морфологических критериев (НГ).
Установлен ряд факторов, оказывающих наиболее значимое влияние на почечный прогноз у больных IgA-нефропатией [3, 4, 6, 12, 17, 18, 21, 30, 34, 45, 46, 54, 55, 70, 74, 78, 84].
Критерии неблагоприятного прогноза IgA-нефропатии:
Клинико-лабораторные
Генетические
Морфологические
Для стандартизации выраженности изменений при световой микроскопии, определения прогноза и выбора тактики лечения была разработана Оксфордская классификация IgA-нефропатии [80, 81]. Показано, что с неблагоприятным почечным прогнозом (независимо от исходных клинический проявлений, а также уровня ПУ и качества контроля АГ) коррелирует выраженность следующих морфологических изменений (шкала MEST):
Однако данная классификация не учитывает наличие полулуний и некротизирующих повреждений.
Оксфордская классификация применяется в Европе, Северной Америке, Китае. В нашей стране доказательная база ее валидности недостаточная, необходимы дальнейшие исследования.
Стратификация групп риска больных IgA-нефропатией:
-
низкий риск: пациенты без ПУ или с ПУ<0,5 г/сут, нормальным АД, нормальной СКФ;
-
умеренный риск: пациенты с ПУ >0,5-1 г/сут и сохранной функцией почек и/или с умеренно сниженной СКФ (но не <50 мл/мин/1,73 м2) и/или с АГ;
-
высокий риск: пациенты с ПУ >3 г/сут и/или с СКФ <30 мл/мин/1,73 м2, пациенты с быстропрогрессирующим гломерулонефритом (БПГН).
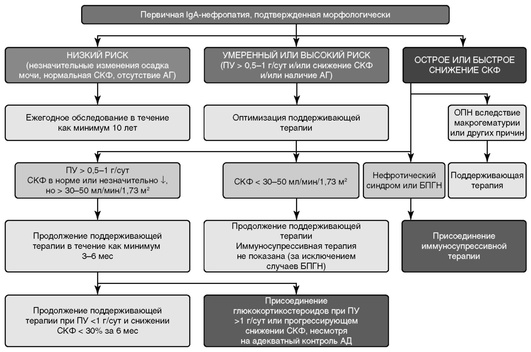
Отбор больных в группы терапии осуществляется в соответствии с выявленным риском прогрессирования, общий алгоритм лечения представлен на рис. 1.
Отбор пациентов в группы терапии
-
Больным с изолированной гематурией, а также с сочетанием гематурии с минимальной ПУ (<0,5 г/сут), нормальной СКФ и отсутствием АГ, лечение не показано. Эти пациенты должны находиться под наблюдением нефролога и раз в 6-12 мес проходить обследование (оценка ПУ, уровня креатинина/СКФ, АД) для своевременного выявления показаний к началу терапии.
-
При персистировании ПУ >0,5 г/сут показана нефропротективная терапия. Возможно присоединение рыбьего жира.
-
При ПУ нефротического уровня или персистировании ПУ >1 г/сут (несмотря на терапию ИАПФ/БРА в течение 3-6 мес. и адекватный контроль АД) и СКФ >50 мл/мин/1,73 м2 показана иммуносупрессивная терапия.
3.1. НЕФРОПРОТЕКТИВНАЯ ТЕРАПИЯ
Нефропротективная терапия [8, 9, 13, 19, 20, 23, 37, 41, 48, 49, 64, 66]
Рекомендация 3.1.1. Предлагается лечение ингибиторами ангиотензин-превращающего фермента (ИАПФ) или БРА при ПУ от 0,5 до 1 г/сут (2D).
Рекомендация 3.1.2. Рекомендуется длительное лечение ИАПФ или блокаторами рецепторов ангиотензина II 1 типа (БРА) при ПУ >1 г/сут, с повышением дозы препаратов в зависимости от АД (1В).
Рекомендация 3.1.3. При IgA-нефропатии целевым следует считать АД <130/80 мм рт.ст. для пациентов с ПУ <1 г/сут и АД <125/75 мм рт.ст. при ПУ >1 г/сут (НГ).
Рекомендация 3.1.4. Предлагается повышение дозы ИАПФ и БРА до максимально переносимых для достижения ПУ<1 г/сут (2С).
Рекомендация 3.1.5. При персистировании ПУ 1 г/сут, несмотря на 3-6-месячную терапию ингибиторами АПФ или БРА и адекватный контроль АД, предлагается использование рыбьего жира (2D).
Рекомендация 3.1.6. У пациентов с IgA-нефропатией и дислипидемией целесообразна коррекция липидных нарушений согласно соответствующим рекомендациям для больных хронической болезнью почек (ХБП) (НГ).
3.2. ИММУНОСУПРЕССИВНАЯ ТЕРАПИЯ
Рекомендация 3.2.1. Пациентам с персистирующей ПУ >1 г/сут (несмотря на 3-6-месячное оптимальное поддерживающее лечение ИАПФ/БРА, адекватный контроль АД) и СКФ > 50 мл/мин/1,73 м2, предлагается проведение 6-месячного курса монотерапии кортикостероидами (2С).
При отсутствии ответа на монотерапию глюкокортикоидами (ГК) тактика дальнейшей иммуносупрессивной терапии не определена, соответствующие рандомизированные клинические исследования (РКИ) не проводились.
Рекомендация 3.2.2. У пациентов с IgA-нефропатией предлагается не применять сочетание кортикостероидов с циклофосфамидом или азатиоприном (за исключением случаев IgA-нефропатии с полулуниями и быстрым снижением функции почек) (2D).
Рекомендация 3.2.3. У больных IgA-нефропатией с СКФ <30 мл/ мин/1,73 м2 предлагается не использовать иммуносупрессивные препараты, за исключением случаев IgA-нефропатии с полулуниями и быстрым снижением функции почек (2С).
У отдельных больных применение ГК в сочетании с циклофосфамидом или азатиоприном снижает активность IgA-нефропатии, однако отсутствует доказательная база влияния данной терапии на отдаленный прогноз.
Рекомендация 3.2.4. Предлагается не использовать микофенолата мофетил для лечения IgA-нефропатии в качестве препарата первой линии (НГ).
Отсутствуют доказательства большей или сопоставимой с преднизолоном эффективности применения других иммуносупрессивных препаратов (в том числе микофенолата мофетила) в качестве терапии первой линии у больных с IgA-нефропатией.
3.3. ЛЕЧЕНИЕ АТИПИЧНЫХ ФОРМ IGA-НЕФРОПАТИИ
Болезнь минимальных изменений с депозитами IgA
Рекомендация 3.3.1. У пациентов с НС и выявленными при биопсии признаками БМИ с мезангиальными депозитами IgA рекомендуется проводить лечение в соответствии с рекомендациями для БМИ (2В).
IgA-нефропатия с макрогематурией и ассоциированной с ней острой почечной недостаточностью
Рекомендация 3.3.2. У пациентов с подтвержденной IgA-нефропатией при развитии острой почечной недостаточности (ОПН), ассоциированной с макрогематурией, целесообразно провести повторную биопсию почки, если через 5 дней после начала снижения почечной функции не наступает улучшение (НГ).
Рекомендация 3.3.3. Предлагается проводить поддерживающую терапию ОПН при IgA-нефропатии, если при биопсии, выполненной во время эпизода макрогематурии, имеются только признаки острого канальцевого некроза с эритроцитарными цилиндрами в просвете канальцев (2С).
IgA-нефропатия с полулуниями
Рекомендация 3.3.4. При IgA-нефропатии с быстропрогрессирующим ухудшением функции и выявлением в биоптате полулуний более чем в 50% клубочков предлагается лечение кортикостероидами и циклофосфамидом по схеме, аналогичной схеме лечения АНЦА-васкулитов (2D).
3.4. ДРУГИЕ ВИДЫ ЛЕЧЕНИЯ
-
Диета. Диетические рекомендации разрабатываются индивидуально с учетом особенностей течения заболевания у конкретного больного в соответствии с рекомендациями по лечению хронической болезнью почек (ХБП):
-
низкоаллергенная диета (ограничение глютена, мяса, молока) не замедляет прогрессирования заболевания, но может быть предложена при сочетании IgA-нефропатии с целиакией (НГ);
-
по мере снижения функции почек при отсутствии противопоказаний ограничивают потребление белка с пищей: до 0,6 г/кг массы тела/сут при СКФ ниже 60 мл/мин/1,73 м2 (при условии применения кето-аналогов аминокислот);
-
больным с ожирением, гиперлипидемией, снижением толерантности к углеводам, лимитируют употребление животных жиров и легко усваиваемых углеводов.
Рекомендация 3.4.1. Целесообразна санация очагов инфекции, провоцирующих обострение заболевания (НГ).
Рекомендация 3.4.2. Предлагается не проводить плановую тонзиллэктомию при IgA-нефропатии (2С). Тонзиллэктомия может быть предложена отдельным больным с частыми рецидивами IgA-нефропатии на фоне обострений хронического тонзиллита, при котором не эффективны консервативные мероприятия (НГ).
Рекомендация 3.4.3. Предлагается не использовать дезагреганты для лечения IgA-нефропатии (2С).
4. Прогноз
Течение IgA-нефропатии считают в целом благоприятным. У больных с минимальной ПУ отмечается низкий риск прогрессирования.
Факторы неблагоприятного прогноза обсуждались выше (см. Рекомендацию. 3).
При выраженной ПУ и/или повышенном уровне креатинина в крови ТПН развивается через 10 лет у 15-25%, через 20 лет - у 20-30% больных [17, 74].
IgA-нефропатия рецидивирует в 20-60% трансплантатов. Рецидив заболевания приводит к ухудшению функции и потере трансплантата в 1,3-16% случаев [11, 58].
СПИСОК ЛИТЕРАТУРЫ
-
Мухин Н.А., Козловская Л.В., Шилов Е.М. и др. Рациональная фармакотерапия в нефрологии : руководство для практикующих врачей. М. : Литтерра, 2006. 896 с.
-
Ballardie F.W., Roberts I.S. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy // J. Am. Soc. Nephrol. 2002. Vol. 13. P. 142-148.
-
Bartosik L.P., Lajoie G., Sugar L., Cattran D.C. Predicting progression in IgA nephropathy // Am. J. Kidney Dis. 2001. Vol. 38. P. 728-735.
-
Berthoux F., Mohey H., Laurent B. et al. Predicting the risk for dialysis or death in IgA nephropathy // J. Am. Soc. Nephrol. 2011. Vol. 22. P. 752-761.
-
Bisceglia L., Cerullo G., Forabosco P. et al. Genetic heterogeneity in Italian families with IgA nephropathy: suggestive linkage for two novel IgA nephropathy loci // Am. J. Hum. Genet. 2006. Vol. 79. P. 1130-1134.
-
Bonnet F., Deprele C., Sassolas A. et al. Excessive body weight as a new independent risk factor for clinical and pathological progression in primary IgA nephritis // Am. J. Kidney Dis. 2001. Vol. 37. P. 720-727.
-
Boyd J.K., Cheung C.K., Molyneux K. et al. An update on the pathogenesis and treatment of IgA nephropathy // Kidney Int. 2012. Vol. 81. P. 833-843.
-
Catapano F., Chiodini P., De Nicola L. et al. Antiproteinuric response to dual blockade of the renin-angiotensin system in primary glomerulonephritis: metaanalysis and metaregression // Am. J. Kidney Dis. 2008. Vol. 52. P. 475-485.
-
Cattran D.C. Is proteinuria reduction by angiotensin-converting enzyme inhibition enough to prove its role in renal protection in IgA nephropathy? // J. Am. Soc. Nephrol. 2007. Vol. 18. P. 1633-1634.
-
Cheng I.K., Chan K.W., Chan M.K. Mesangial IgA nephropathy with steroid-responsive nephrotic syndrome: disappearance of mesangial IgA deposits following steroid-induced remission // Am. J. Kidney Dis. 1989. Vol. 14. P. 361-364.
-
Choy B.Y., Chan T.M., Lai K.N. Recurrent glomerulonephritis after kidney transplantation // Am. J. Transplant. 2006. Vol. 6. P. 2535-2542.
-
Coppo R., D’Amico G. Factors predicting progression of IgA nephropathies // J. Nephrol. 2005. Vol. 18. P. 503-512.
-
Coppo R., Peruzzi L., Amore A. et al. IgACE: a placebo-controlled, randomized trial of angiotensin-converting enzyme inhibitors in children and young people with IgA nephropathy and moderate proteinuria // J. Am. Soc. Nephrol. 2007. Vol. 18. P. 1880-1888.
-
Coppo R., Roccatello D., Amore A. et al. Effects of a gluten-free diet in primary IgA nephropathy // Clin. Nephrol. 1990. Vol. 33. P. 72-86.
-
D’Amico G., Colasanti G., Barbiano di Belgioioso G. et al. Long-term follow-up of IgA mesangial nephropathy: clinico-histological study in 374 patients // Semin. Nephrol. 1987. Vol. 7. P. 355-358.
-
Dal Canton A., Amore A., Barbano G. et al. One-year angiotensin-con-verting enzyme inhibition plus mycophenolate mofetil immunosuppression in the course of early IgA nephropathy: a multicenter, randomised, controlled study // J. Nephrol. 2005. Vol. 18. P. 136-140.
-
D’Amico G. Influence of clinical and histological features on actuarial renal survival in adult patients with idiopathic IgA nephropathy, membranous nephropa-thy, and membranoproliferative glomerulonephritis: survey of the recent literature // Am. J. Kidney Dis. 1992. Vol. 20. P. 315-323.
-
Daniel L., Saingra Y., Giorgi R. et al. Tubular lesions determine prognosis of IgA nephropathy // Am. J. Kidney Dis. 2000. Vol. 35. P. 13-20.
-
Donadio J.V. Jr, Bergstralh E.J., Offord K.P. et al. A controlled trial of fish oil in IgA nephropathy. Mayo Nephrology Collaborative Group // N. Engl. J. Med. 1994. Vol. 331. P. 1194-1199.
-
Donadio J.V. Jr, Larson T.S., Bergstralh E.J., Grande J.P. A randomized trial of high-dose compared with low-dose omega-3 fatty acids in severe IgA ne-phropathy // J. Am. Soc. Nephrol. 2001. Vol. 12. P. 791-799.
-
Donadio J.V., Bergstralh E.J., Grande J.P., Rademcher D.M. Proteinuria patterns and their association with subsequent end-stage renal disease in IgA nephropathy // Nephrol. Dial. Transplant. 2002. Vol. 17. P. 1197-1203.
-
Donadio J.V., Grande J.P. IgA nephropathy // N. Engl. J. Med. 2002. Vol. 347. P. 738-748.
-
Ferraro P.M., Ferraccioli G.F., Gambaro G. et al. Combined treatment with renin-angiotensin system blockers and polyunsaturated fatty acids in proteinuric IgA nephropathy: a randomized controlled trial // Nephrol. Dial. Transplant. 2009. Vol. 24. P. 156-160.
-
Ferri C., Puccini R., Longombardo G. et al. Low-antigen-content diet in the treatment of patients with IgA nephropathy // Nephrol. Dial. Transplant. 1993. Vol. 8. P. 1193-1198.
-
Floege J., Eitner F. Combined immunosuppression in high-risk patients with IgA nephropathy? // J. Am. Soc. Nephrol. 2010. Vol. 21. P. 1604-1606.
-
Floege J., Eitner F. Current therapy for IgA nephropathy // J. Am. Soc. Nephrol. 2011. Vol. 22. P. 1785-1794.
-
Floege J., Eitner F. Present and future therapy options in IgA-nephropa-thy // J. Nephrol. 2005. Vol. 18. P. 354-361.
-
Gharavi A.G., Yan Y., Scolari F. et al. IgA nephropathy, the most common cause of glomerulonephritis, is linked to 6q22-23 // Nat. Genet. 2000. Vol. 26. P. 354-357.
-
Habib R., Niaudet P., Levy M. Schmnlein-Henoch purpura nephritis and IgA nephropathy // Renal Pathology / eds C. Tisher, B.M. Brenner. Philadelphia : J.B. Lippincott, 1994. P. 472-523.
-
Harden P.N., Geddes C., Rowe P.A. et al. Polymorphisms in angiotensin-converting-enzyme gene and progression of IgA nephropathy // Lancet. 1995. Vol. 345. P. 1540-1542.
-
UTL: http://www.uptodate.com/contents/treatment-and-prognosis-of-iga-ephropathy?source=related_link
-
Hunley T.E., Julian B.A., Phillips J.A. 3rd. et al. Angiotensin converting enzyme gene polymorphism: potential silencer motif and impact on progression in IgA nephropathy // Kidney Int. 1996. Vol. 49. P. 571-577.
-
Ibels L.S., Gyory A.Z. IgA nephropathy: analysis of the natural history, important factors in the progression of renal disease, and a review of the literature // Medicine (Baltimore). 1994. Vol. 73. P. 79-102.
-
Ikee R., Kobayashi S., Saigusa T. et al. Impact of hypertension and hypertension-related vascular lesions in IgA nephropathy // Hyperrtens Res. 2006. Vol. 29. P. 15-22.
-
Jennette J.C., Wall S.D., Wilkman A.S. Low incidence of IgA nephropathy in blacks // Kidney Int. 1985. Vol. 28. P. 944-950.
-
Kamei K., Nakanishi K., Ito S. et al. Long-term results of a randomized controlled trial in childhood IgA nephropathy // Clin. J. Am. Soc. Nephrol. 2011. Vol. 6. P. 1301-1307.
-
Kanno Y., Okada H., Saruta T., Suzuki H. Blood pressure reduction associated with preservation of renal function in hypertensive patients with IgA nephropathy: a 3-year follow-up // Clin. Nephrol. 2000. Vol. 54. P. 360-365.
-
Kawamura T., Yoshimura M., Miyazaki Y. et al. A multicenter randomized controlled trial of tonsillectomy combined with steroid pulse therapy in patients with immunoglobulin A nephropathy // Nephrol. Dial. Transplant. 2014. Vol. 29. P. 1546-1553.
-
Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulone-phritis Work Group. KDIGO Clinical Practice Guideline for Glomerulonephritis. diapter 10: immunoglobulin A nephropathy // Kidney Int. Suppl. 2012. Vol. 2. P. S209-S217.
-
Kim Y.C., Chin H.J., Koo H.S., Kim S. Tacrolimus decreases albumin-uria in patients with IgA nephropathy and normal blood pressure: a double-blind randomized controlled trial of efficacy of tacrolimus on IgA nephropathy // PLoS One. 2013. Vol. 8. Article ID e71545.
-
Kunz R., Friedrich C., Wolbers M., Mann J.F. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease // Ann. Intern. Med. 2008. Vol. 148. P. 30-48.
-
Lai K.N., Lai F.M., Ho C.P., Chan K.W. Corticosteroid therapy in IgA nephropathy with nephrotic syndrome: a long-term controlled trial // Clin. Nephrol. 1986. Vol. 26. P. 174-180.
-
Lai K.N., Lai F.M., Leung A.C. et al. Plasma exchange in patients with rapidly progressive idiopathic IgA nephropathy: a report of two cases and review of literature // Am. J. Kidney Dis. 1987. Vol. 10. P. 66-70.
-
Lai K.N., Lai F.M., Li P.K., Vallance-Owen J. Cyclosporin treatment of IgA nephropathy: a short term controlled trial // Br. Med. J. (Clin. Res. Ed.). 1987. Vol. 295. P. 1165-1168.
-
Le W., Liang S., Hu Y. et al. Long-term renal survival and related risk factors in patients with IgA nephropathy: results from a cohort of 1155 cases in a Chinese adult population // Nephrol. Dial. Transplant. 2012. Vol. 27. P. 1479-1485.
-
Lee H.S., Lee M.S., Lee S.M. et al. Histological grading of IgA nephropa-thy predicting renal outcome: revisiting H. S. Lee’s glomerular grading system // Nephrol. Dial. Transplant. 2005. Vol. 20. P. 342-348.
-
Li P.K., Ho K.K., Szeto C.C. et al. Prognostic indicators of IgA nephropathy in the Chinese - clinical and pathological perspectives // Nephrol. Dial. Transplant. 2002. Vol. 17. P. 64-69.
-
Li P.K., Kwan B.C., Chow K.M. et al. Treatment of early immunoglobulin A nephropathy by angiotensin-converting enzyme inhibitor // Am. J. Med. 2013. Vol. 126. P. 162-168.
-
Li P.K., Leung C.B., Chow K.M. et al. Hong Kong study using valsartan in IgA nephropathy (HKVIN): a double-blind, randomized, placebo-controlled study // Am. J. Kidney Dis. 2006. Vol. 47. P. 751-760.
-
Locatelli F., Del Vecchio L., Pozzi C. The patient with IgA glomerulone-phritis-what is the role of steroid treatment? // Nephrol. Dial. Transplant. 1999. Vol. 14. P. 1057-1060.
-
Lv J., Xu D., Perkovic V. et al. Corticosteroid therapy in IgA nephropathy // J. Am. Soc. Nephrol. 2012. Vol. 23. P. 1108-1114.
-
Maes B.D., Oyen R., Claes K. et al. Mycophenolate mofetil in IgA nephropathy: results of a 3-year prospective placebo-controlled randomized study // Kidney Int. 2004. Vol. 65. P. 1842-1849.
-
Maisonneuve P., Agodoa L., Gellert R. et al. Distribution of primary renal diseases leading to end-stage renal failure in the United States, Europe, and Australia/New Zealand: results from an international comparative study // Am. J. Kidney Dis. 2000. Vol. 35. P. 157-165.
-
Maixnerova D., Bauerova L., Skibova J. et al. The retrospective analysis of 343 Czech patients with IgA nephropathy - one centre experience // Nephrol. Dial. Transplant. 2012. Vol. 27. P. 1492-1498.
-
Manno C., Strippoli G.F., D’Altri C. et al. A novel simpler histological classification for renal survival in IgA nephropathy: a retrospective study // Am. J. Kidney Dis. 2007. Vol. 49. P. 763-775.
-
Manno C., Torres D.D., Rossini M. et al. Randomized controlled clinical trial of corticosteroids plus ACE-inhibitors with long-term follow-up in proteinuric IgA nephropathy // Nephrol. Dial. Transplant. 2009. Vol. 24. P. 3694-3701.
-
McIntyre C.W., Fluck R.J., Lambie S.H. Steroid and cyclophosphamide therapy for IgA nephropathy associated with crescenteric change: an effective treatment // Clin. Nephrol. 2001. Vol. 56. P. 193-198.
-
Moroni G., Longhi S., Quaglini S. et al. The long-term outcome of renal transplantation of IgA nephropathy and the impact of recurrence on graft survival // Nephrol. Dial. Transplant. 2013. Vol. 28. P. 1305-1314.
-
Mustonen J., Pasternack A., Rantala I. The nephrotic syndrome in IgA glomerulonephritis: response to corticosteroid therapy // Clin. Nephrol. 1983. Vol. 20. P. 172-176.
-
Nair R., Walker P.D. Is IgA nephropathy the commonest primary glomerulopathy among young adults in the USA? // Kidney Int. 2006. Vol. 69. P. 1455-1458.
-
Novak J., Julian B.A., Mestecky J., Renfrow M.B. Glycosylation of IgA1 and pathogenesis of IgA nephropathy // Semin. Immunopathol. 2012. Vol. 34. P. 365-382.
-
Pozzi C., Andrulli S., Del Vecchio L. et al. Corticosteroid effectiveness in IgA nephropathy: long-term results of a randomized, controlled trial // J. Am. Soc. Nephrol. 2004. Vol. 15. P. 157-163.
-
Pozzi C., Andrulli S., Pani A. et al. Addition of azathioprine to corticosteroids does not benefit patients with IgA nephropathy // J. Am. Soc. Nephrol. 2010. Vol. 21. P. 1783-1790.
-
Praga M., Gutierrez E., Gonzalez E. et al. Treatment of IgA nephropathy with ACE inhibitors: a randomized and controlled trial // J. Am. Soc. Nephrol. 2003. Vol. 14. P. 1578-1583.
-
Rasche F.M., Schwarz A., Keller F. Tonsillectomy does not prevent a progressive course in IgA nephropathy // Clin. Nephrol. 1999. Vol. 51. P. 147-152.
-
Reid S., Cawthon P.M., Craig J.C. et al. Non-immunosuppressive treatment for IgA nephropathy // Cochrane Database Syst. Rev. 2011. Issue 3: CD003962.
-
Roccatello D., Ferro M., Coppo R. et al. Report on intensive treatment of extracapillary glomerulonephritis with focus on crescentic IgA nephropathy // Nephrol. Dial. Transplant. 1995. Vol. 10. P. 2054-2059.
-
Suzuki H., Kiryluk K., Novak J. et al. The pathophysiology of IgA nephropathy // J. Am. Soc. Nephrol. 2011. Vol. 22. P. 1795-803.
-
Suzuki K., Honda K., Tanabe K. et al. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan // Kidney Int. 2003. Vol. 63. P. 2286-2294.
-
Syrjanen J., Mustonen J., Pasternack A. Hypertriglyceridaemia and hyper-uricaemia are risk factors for progression of IgA 48. nephropathy // Nephrol. Dial. Transplant. 2000. Vol. 15. P. 34-42.
-
Szeto C.C., Lai F.M., To K.F. et al. The natural history of immunoglobu-lin a nephropathy among patients with hematuria and minimal proteinuria // Am. J. Med. 2001. Vol. 110. P. 434-437.
-
Tang S.C., Tang A.W., Wong S.S. et al. Long-term study of mycophenolate mofetil treatment in IgA nephropathy // Kidney Int. 2010. Vol. 77. P. 543-549.
-
Tumlin J.A., Lohavichan V., Hennigar R. Crescentic, proliferative IgA ne-phropathy: clinical and histological response to methylprednisolone and intravenous cyclophosphamide // Nephrol. Dial. Transplant. 2003. Vol. 18. P. 1321-1329.
-
Wakai K., Kawamura T., Endoh M. et al. A scoring system to predict renal outcome in IgA nephropathy: from a nationwide prospective study // Nephrol. Dial. Transplant. 2006. Vol. 21. P. 2800-2808.
-
Waldherr R., Rambausek M., Duncker W.D., Ritz E. Frequency of mesan-gial IgA deposits in a non-selected autopsy series // Nephrol. Dial. Transplant. 1989. Vol. 4. P. 943-946.
-
Walker R.G., Yu S.H., Owen J.E., Kincaid-Smith P. The treatment of mesangial IgA nephropathy with cyclophosphamide, dipyridamole and warfarin: a two-year prospective trial // Clin. Nephrol. 1990. Vol. 34. P. 103-107.
-
Wang Y., Chen J., Wang Y. et al. A meta-analysis of the clinical remission rate and long-term efficacy of tonsillectomy in patients with IgA nephropathy // Nephrol. Dial. Transplant. 2011. Vol. 26. P. 1923-1931.
-
Weber C.L., Rose C.L., Magil A.B. Focal segmental glomerulosclerosis in mild IgA nephropathy: a clinical-pathologic study // Nephrol. Dial. Transplant. 2009. Vol. 24. P. 483-488.
-
Welch T.R., McAdams A.J., Berry A. Rapidly progressive IgA nephropathy // Am. J. Dis. Child. 1988. Vol. 142. P. 789-793.
-
Working Group of the International IgA Nephropathy Network and the Renal Pathology Society, Cattran D.C., Coppo R. et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification // Kidney Int. 2009. Vol. 76. P. 534-545.
-
Working Group of the International IgA Nephropathy Network and the Renal Pathology Society, Roberts I.S., Cook H.T. et al. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility // Kidney Int. 2009. Vol. 76. P. 546-556.
-
Wyatt R.J., Julian B.A. IgA nephropathy // N. Engl. J. Med. 2013. Vol. 368. P. 2402-2414.
-
Xie Y., Nishi S., Ueno M. et al. The efficacy of tonsillectomy on long-term renal survival in patients with IgA nephropathy // Kidney Int. 2003. Vol. 63. P. 1861-1867.
-
Yamamoto R., Nagasawa Y., Shoji T. et al. Cigarette smoking and progression of IgA nephropathy // Am. J. Kidney Dis. 2010. Vol. 56. P. 313-324.
-
Yoshikawa N., Honda M., Iijima K. et al. Steroid treatment for severe childhood IgA nephropathy: a randomized, controlled trial // Clin. J. Am. Soc. Nephrol. 2006. Vol. 1. P. 511-517.
-
Zand L., Fervenza F.C. Does tonsillectomy have a role in the treatment of patients with immunoglobulin A nephropathy? // Nephrol. Dial. Transplant. 2014. Vol. 29. P. 1456-1459.
ЭКСТРАКАПИЛЛЯРНЫЙ ГЛОМЕРУЛОНЕФРИТ С ПОЛУЛУНИЯМИ
1. Определение, эпидемиология, этиология
Быстропрогрессирующий ГН (БПГН) - ургентная нефрологическая ситуация, требующая срочных диагностических и лечебных мероприятий. БПГН клинически характеризуется остронефритическим синдромом с быстро прогрессирующей почечной недостаточностью (ПН) (удвоение креатинина в срок 3 мес), морфологически - наличием более чем в 50% клубочков экстракапиллярных клеточных или фиброзно-клеточных полулуний.
Синонимы термина: подострый гломерулонефрит (ГН), злокачественный ГН. Общепринятый морфологический термин, используемый для обозначения БПГН, - экстракапиллярный ГН с полулуниями.
Эпидемиология
Частота БПГН составляет 2-10% всех форм ГН, регистрируемых в специализированных нефрологических стационарах.
Этиология
БПГН может быть идиопатическим или развиваться в рамках системных заболеваний (АНЦА-ассоциированный васкулит, синдром Гудпасчера, СКВ).
2. Патогенез
Полулуния являются следствием выраженного повреждения клубочков с разрывом стенок капилляров и проникновением плазменных белков и воспалительных клеток в пространство капсулы Шумлянского-Боумена.
Основной причиной такого тяжелого повреждения является воздействие АНЦА, анти-БМК-АТ и ИК. Клеточный состав полулуний представлен в основном пролиферирующими париетальными эпителиальными клетками и макрофагами. Эволюция полулуний - обратное развитие или фиброз - зависит от степени накопления макрофагов в пространстве капсулы Шумлянского-Боумена и ее структурной целостности. Преобладание в клеточных полулуниях макрофагов сопровождается разрывом капсулы, последующим поступлением из интерстиция фибробластов и миофибробластов, синтезом этими клетками матриксных белков - коллагенов I и III типов, фибронектина, что ведет к необратимому фиброзу полулуний. Важная роль в регуляции процессов привлечения и накопления макрофагов в полулуниях принадлежит хемокинам - моноцитарному хемоаттрактантному протеину-1 и макрофагальному воспалительному протеину-1. Высокая экспрессия этих хемокинов в местах формирования полулуний с высоким содержанием макрофагов обнаруживается при БПГН с наиболее тяжелым течением и плохим прогнозом. Важным фактором, приводящим к фиброзу полулуний, является фибрин, в который трансформируется фибриноген, попадающий в полость капсулы вследствие некроза капиллярных петель клубочка.
3. Классификация
В зависимости от преимущественного механизма повреждения, клинической картины и лабораторных показателей в настоящее время выделены 5 иммунопатогенетических типов БПГН (Glassock, 1997). Основными иммунопатологическими критериями, определяющими каждый тип БПГН, являются тип свечения иммунореактантов в почечном биоптате и наличие повреждающего фактора (АТ к БМК, ИК, АНЦА) в сыворотке больного (табл. 1).
Патогенетический тип ЭКГН |
ИФ-микроскопия почечной ткани (тип свечения) |
Сыворотка |
||
|---|---|---|---|---|
анти-БМК |
комплемент (снижение уровня) |
АНЦА |
||
Тип I |
Линейное |
+ |
- |
- |
Тип II |
Гранулярное |
- |
+ |
- |
Тип III |
- |
- |
+ |
|
Тип IV |
Линейное |
+ |
- |
+ |
Тип V |
- |
- |
- |
- |
Тип I (антительный, анти-БМК-нефрит) обусловлен повреждающим действием АТ к БМК. Характеризуется линейным свечением АТ в почечном биоптате и наличием циркулирующих АТ к БМК в сыворотке крови. Существует или как изолированная (идиопатическая) болезнь почек, или как заболевание с содружественным поражением легких и почек (синдром Гудпасчера).
Тип II (иммунокомплексный) вызван депозитами ИК в различных отделах почечных клубочков (в мезангии и капиллярной стенке). В почечном биоптате выявляется в основном гранулярный тип свечения, в сыворотке анти-БМК-АТ и АНЦА отсутствуют, у многих больных может быть снижен уровень комплемента. Наиболее характерен для БПГН, связанных с инфекциями (постстрептококковый БПГН), КГ, СКВ.
Тип III (малоиммунный). Повреждение обусловлено клеточными иммунными реакциями, в том числе нейтрофилами и моноцитами, активированными антинейтрофильными цитоплазматическими АТ (АНЦА). Свечение иммуноглобулинов и комплемента в биоптате отсутствует или незначительно (pauci-immune, малоиммунный ГН), в сыворотке выявляются АНЦА, направленные против протеиназы-3 или миелопероксидазы (МПО). Этот тип ЭКГН - проявление АНЦА-ассоциированного васкулита [микроскопический полиангиит (МПА), гранулематоз с полиангиитом (Вегенера) (ГПА)].
Тип IV представляет собой сочетание двух патогенетических типов - антительного (первый тип) и АНЦА-ассоциированного, или малоиммунного (третий тип). При этом в сыворотке крови определяются и АТ к БМК, и АНЦА, а в почечном биоптате выявляют линейное свечение АТ к БМК как при классическом анти-БМК-нефрите. При этом возможна пролиферация мезангиальных клеток, отсутствующая при классическом АТ-типе ЭКГН.
Тип V (истинный идиопатический). При этом крайне редком типе иммунные факторы повреждения не удается выявить ни в циркуляции (отсутствуют анти-БМК-АТ и АНЦА, уровень комплемента нормальный), ни в почечном биоптате (полностью отсутствует свечение иммуноглобулинов). Предполагается, что в его основе лежит клеточный механизм повреждения почечной ткани.
Среди всех типов БПГН более половины (55%) приходится на АНЦА-ассоциированный БПГН (III тип), два других типа БПГН (I и II) распределяются примерно поровну (20% и 25%). Характеристика основных типов БПГН представлена в табл. 2.
| Тип БПГН | Характеристика | Клинические варианты | Частота, % |
|---|---|---|---|
I |
Опосредованный АТ к БМК: линейные отложения IgG при иммуногистологическом исследовании ткани почек |
Синдром Гудпасчера. Изолированное поражение почек, ассоциированное с АТ к БМК |
5 |
II |
Иммунокомплексный: гранулярные отложения иммуноглобулина в клубочках почки |
Постинфекционный. Постстрептококковый. При висцеральных абсцессах. Люпус-нефрит. Геморрагический васкулит. IgA-нефропатия. Смешанная КГ. МБПГН |
30-40 |
III |
АНЦА-ассоциированный: малоиммунный с отсутствием иммунных отложений при иммунологическом исследовании |
ГПА. МПА. Эозинофильный гранулематоз с полиангиитом (ЭГПА) |
50 |
IV |
Сочетание I и III типов |
- |
- |
V |
АНЦА-негативный васкулит почек: с отсутствием иммунных отложений |
Идиопатический |
5-10 |
По наличию тех или иных серологических маркеров (и их комбинации) можно предположить тип свечения в почечном биоптате и, соответственно, механизм повреждения - патогенетический тип БПГН, что важно учитывать при выборе программы лечения.
Рекомендация 1. Во всех случаях БПГН необходимо выполнять биопсию почки, по возможности безотлагательно. Морфологическое исследование ткани почки должно проводиться с обязательным применением люминесцентной микроскопии.
Комментарий. АНЦА-ассоциированный системный васкулит (АНЦА-СВ) - наиболее частая причина БПГН. Вовлечение почек при этих заболеваниях является фактором неблагоприятного прогноза в отношении как почечной, так и общей выживаемости. В связи с этим биопсия почки чрезвычайно важна не только с диагностической, но и с прогностической точки зрения.
4. Клинические проявления
Клинический синдром БПГН включает два компонента:
Таким темпам прогрессирования соответствует удвоение уровня креатинина сыворотки за каждые 3 мес болезни. Однако часто фатальная потеря функции происходит всего за несколько (1-2) недель, что соответствует критериям ОПН.
5. Принципы диагностики
БПГН диагностируют на основании оценки темпа ухудшения функции почек и выделения ведущего нефрологического синдрома (остронефритический и/или нефротический).
5.1. ЛАБОРАТОРНАЯ ДИАГНОСТИКА БЫСТРОПРОГРЕССИРУЮЩЕГО ГЛОМЕРУЛОНЕФРИТА
-
Общий анализ крови: нормохромная анемия, возможны нейтрофильный лейкоцитоз или лейкопения, тромбоцитоз или тромбоцитопения, повышение скорости оседания эритроцитов.
-
Общий анализ мочи: ПУ (от минимальной до массивной), эритроцитурия, как правило, выраженная, наличие эритроцитарных цилиндров, лейкоцитурия.
-
Биохимический анализ крови: повышение концентрации креатинина, мочевой кислоты, калия, гипопротеин- и гипоальбуминемия, дислипидемия в случаях НС.
-
Снижение СКФ (определенное по клиренсу креатинина - проба Реберга и/или расчетными методами CKD-EPI, MDRD; использование формулы Кокрофта-Голта нежелательно в связи с завышением СКФ на 20-30 мл.
5.2. ГИСТОЛОГИЧЕСКИЕ ИССЛЕДОВАНИЯ БИОПТАТА ПОЧКИ
Комментарий. Всем больным БПГН показана биопсия почки. Проведение ее необходимо в первую очередь с целью оценки прогноза и выбора оптимального метода лечения: своевременно примененная агрессивная схема иммуносупрессивной терапии иногда позволяет добиться восстановления фильтрационной функции почек даже в ситуации, когда степень ее ухудшения достигла ТПН. В связи с этим при БПГН биопсию почки необходимо выполнять и при выраженной, требующей проведения ГД ПН.
Морфологическую характеристику разных типов БПГН см. в рекомендациях по анти-БМК ГН, АНЦА-ассоциированному ГН (АНЦА-ГН) и волчаночному нефриту.
5.3. ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
При выявлении синдрома БПГН необходимо исключить состояния, которые внешне напоминают (имитируют) БПГН, но имеют другую природу и поэтому требуют иного терапевтического подхода. По своей природе это три группы заболеваний:
-
нефриты - острый постинфекционный и острый интерстициальный, как правило, с благоприятным прогнозом, при которых только в некоторых случаях применяются иммуносупрессанты;
-
острый канальцевый некроз со своими закономерностями течения и лечения;
-
группа сосудистых болезней почек, объединяющих поражение сосудов разного калибра и различной природы (тромбозы и эмболии крупных сосудов почек, склеродермическую нефропатию, ТМА разного генеза). В большинстве случаев эти состояния можно исключить клинически (табл. 3).
В то же время наличие и особенности внепочечной симптоматики могут указать на болезнь, при которой часто развивается БПГН (СКВ, системные васкулиты, лекарственная реакция).
| Состояния, воспроизводящие БПГН | Отличительные особенности |
|---|---|
АФС-нефропатия |
Наличие сывороточных АТ к кардиолипину классов IgM и IgG и/или АТ к β2-гликопротеиду-1, волчаночного антикоагулянта. Повышение плазменной концентрации D-димера, продуктов деградации фибрина. Отсутствие или незначительные изменения анализа мочи (обычно следовая ПУ, скудный мочевой осадок) при выраженном снижении СКФ |
Клинические проявления артериальных (острый коронарный синдром/острый инфаркт миокарда, острое нарушение мозгового кровообращения) и венозных (тромбоз глубоких вен голеней, тромбоэмболия легочных артерий, тромбоз почечных вен) сосудов, сетчатое ливедо |
|
Гемолитико-уремический синдром |
Связь с инфекционной диареей (при типичном гемолитико-уремическом синдроме). Выявление триггеров активации комплемента (вирусные и бактериальные инфекции, травма, беременность, лекарства). Тяжелая анемия с признаками микроангиопатического гемолиза (повышение уровня ЛДГ, снижение гаптоглобина, шизоцитоз), тромбоцитопения |
Склеродермическая нефропатия |
Кожные и органные признаки системной склеродермии. Выраженный и некупируемый подъем АД. Отсутствие изменений в анализах мочи |
Острый канальцевый некроз |
Связь с приемом лекарственного препарата (особенно НПВП, ненаркотического анальгетика, антибиотика). Макрогематурия (возможно отхождение сгустков крови). Быстрое развитие олигурии |
Острый тубулоинтерстициальный нефрит |
Как правило, четкая причина (прием лекарственного препарата, саркоидоз). Снижение относительной плотности мочи при отсутствии выраженной ПУ |
Холестериновая эмболия внутрипочечных артерий и артериол* |
Связь с эндоваскулярной процедурой, тромболизисом, тупой травмой живота. Выраженный подъем АД. Признаки острофазового ответа [лихорадка, потеря аппетита, массы тела, артралгии, увеличение скорости оседания эритроцитов, сывороточной концентрации С-реактивного белка (СРБ)]. Гиперэозинофилия, эозинофилурия. Сетчатое ливедо с трофическими язвами (чаще на коже нижних конечностей). Системные признаки холестериновой эмболии (внезапная односторонняя слепота, острый панкреатит, гангрена кишки) |
* В редких случаях приводит к развитию БПГН, в том числе АНЦА-ассоциированного
6. Лечение
6.1. ОБЩИЕ ПРИНЦИПЫ ЛЕЧЕНИЯ БЫСТРОПРОГРЕССИРУЮЩЕГО ГЛОМЕРУЛОНЕФРИТА (ЭКСТРАКАПИЛЛЯРНОГО ГЛОМЕРУЛОНЕФРИТА)
-
БПГН встречается чаще как проявление системного заболевания (СКВ, системные васкулиты, эссенциальная смешанная КГ и др.), реже - как идиопатическая болезнь, однако принципы лечения общие.
-
Необходимо, по возможности, экстренное исследование сыворотки на наличие анти-БМК-АТ и АНЦА; биопсия почки необходима для своевременной постановки диагноза (выявления ЭКГН и типа свечения АТ - линейного, гранулярного, малоиммунного), оценки прогноза и выбора тактики терапии.
Рекомендация 1. Для предотвращения необратимой катастрофической потери почечной функции необходимо срочно начинать и сразу же после установления клинического диагноза БПГН (остронефритического синдрома в сочетании с быстропрогрессирующей ПН при нормальных размерах почек и исключении других причин ОПН) (1В).
Комментарии. Задержка лечения на несколько дней может ухудшить эффективность лечения, так как при развитии анурии лечение почти всегда безуспешно. Это единственная форма ГН, при которой опасность развития побочных явлений иммуносупрессивной терапии не сопоставима с возможностью неблагоприятного прогноза при естественном течении болезни и несвоевременном начале лечения.
Рекомендация 1.1. Лечение БПГН следует начинать еще до получения результатов диагностических исследований (серологических, морфологического) с пульс-терапии метилпреднизолоном в дозе до 1000 мг в течение 1-3 дней (1A).
Комментарии.
-
Такая тактика полностью оправдана даже в случае невозможности проведения биопсии почки у больных, тяжесть состояния которых препятствует этой процедуре. Сразу после верификации диагноза БПГН к ГК следует добавить алкилирующие препараты (циклофосфамид в сверхвысоких дозах), особенно у больных с васкулитом (локальнопочечным или системным) и циркулирующими АНЦА и ВН. Интенсивный плазмаферез (ПФ) целесообразно сочетать с иммуносупрессантами в случаях:
-
анти-БМК нефрита при условии начала лечения до появления потребности в ГД;
-
у больных с не анти-БМК ЭКГН, имеющих признаки ПН, требующей лечения ГД в момент постановки диагноза (СКр >500 мкмоль/л) в отсутствие признаков необратимого повреждения почек по данным нефробиопсии (>50% клеточных или фиброзно-клеточных полулуний).
-
Инициальная терапия БПГН зависит от его иммунопатогенетического типа и потребности в диализе в момент постановки диагноза (табл. 4).
Тип |
Серология |
Терапия/потребность в ГД |
|
|---|---|---|---|
нет |
есть |
||
I |
Анти-БМК-болезнь (а-БМK+) (АНЦА-) |
ГК (0,5-1 мг/кг внутрь ± пульс терапия в дозе до 1000 мг в течение 1-3 дней) плазмаферез (интенсивный) |
Консервативное ведение |
II |
ИК-болезнь (а-БМК-), (АНЦА-) |
ГК (внутрь или «пульсы») ± цитостатики (циклофосфамид) - внутрь (2 мг/кг/сут) или внутривенно (15 мг/кг, но не >1 г) |
|
III |
«Малоиммунный» (а-БМК-) (АНЦА+) |
ГК (внутрь или «пульсы») циклофосфамид |
ГС (внутрь или «пульсы») циклофосфамид. Интенсивный ПО - ежедневно в течение 14 дней с объемом замещения 50 мл/кг/сут |
IV |
Комбинированный (а-БМK+) (АНЦА+) |
Как при I типе |
Как при I типе |
V |
«Идиопатический» (а-БМК-) (АНЦА-) |
Как при III типе |
Как при III типе |
Примечание: ГД - гемодиализ.
6.2. РЕКОМЕНДАЦИИ ПО ЛЕЧЕНИЮ ОТДЕЛЬНЫХ ПАТОГЕНЕТИЧЕСКИХ ТИПОВ ЭКГН
6.2.1. Анти-БМК-нефрит (тип I по R. Glassock, 1997), в том числе синдром Гудпасчера
Рекомендация 2. Всем пациентам с анти-БМК ГН (за исключением диализ-зависимых на момент установки диагноза, имеющих 100% полулуний по данным адекватной нефробиопсии и не имеющих при этом легочных кровотечений) следует начинать иммуносупрессию циклофосфамидом, кортикостероидами и плазмаферезом (1B).
Комментарий.
-
При уровне креатинина крови <600 мкмоль/л назначают преднизолон внутрь в дозе 1 мг/кг/сут и циклофосфамид в дозе 2-3 мг/кг/сут. По достижении стабильного клинического эффекта дозу преднизолона постепенно снижают в течение последующих 12 нед, а циклофосфамид полностью отменяют через 10 нед лечения. Терапию иммуносупрессивными препаратами сочетают с интенсивным плазмаферезом, который проводят ежедневно. В случае риска развития легочного кровотечения часть объема удаленной плазмы замещают СЗП. Стабильный эффект достигают после проведения 10-14 сеансов плазмафереза. Такой режим терапии позволяет добиться улучшения функции почек почти у 80% больных, причем снижение азотемии начинается уже через несколько суток после начала плазмафереза.
-
При содержании креатинина в крови >600 мкмоль/л агрессивная терапия малоэффективна и улучшение функции почек возможно лишь у небольшого числа больных с недавней историей заболевания, бурным прогрессированием (в течение 1-2 нед) и наличием в биоптате почки потенциально обратимых изменений. В этих ситуациях основную терапию проводят в сочетании с сеансами ГД.
Рекомендация 3. В случае развития обострений синдрома Гудпасчера применяют тот же терапевтический режим, что и в дебюте болезни.
Рекомендация 4. Проведение поддерживающей иммуносупрессивной терапии при анти-БМК Гн нецелесообразно (1D).
6.2.2. Иммунокомплексный быстропрогрессирующий гломерулонефрит (тип II по R.Glassock, 1997)
Рекомендация 5. При иммунокомплексном БПГН лечение следует начинать с внутривенных «пульсов» метилпреднизолоном (по 1000 мг в течение 3-5 дней) с дальнейшим приемом преднизолона внутрь (60 мг/кг в сутки).
Рекомендация 6. При быстропрогрессирующем волчаночном ГН (IV тип) рекомендовано назначать циклофосфамид (1B) внутривенно в дозе 500 мг каждые 2 нед в течение 3 мес (суммарная доза 3 г) или препараты МФК [микофенолата мофетил (1B) в целевой дозе 3 г/сут в течение 6 мес, или микофенолат натрия в эквивалентной дозе] в сочетании с ГК в виде внутривенных «пульсов» метилпреднизолона в дозе 500-750 мг в течение 3 последовательных дней, и затем преднизолона внутрь 1,00,5 мг/кг/сут в течение 4 нед с постепенным снижением до ≤10 мг/сут к 4-6 мес (1A).
Комментарии. Цитостатики эффективны как при СКВ, так и при криоглобулинемическом ГН (после исключения гепатита, вызванного HCV). При инфицировании HCV показано добавление интерферона альфа.
Рекомендация 7. Лечение плазмаферезом при иммунокомплексном БПГН не показано (2В).
Комментарий. Польза плазмафереза доказана лишь при БПГН у больных с КГ.
6.2.3. Малоиммунный быстропрогрессирующий гломерулонефрит, ассоциированный с антинейтрофильными цитоплазматическими антителами (тип III по R. Glassock, 1997)
Рекомендация 8. В качестве инициальной терапии АНЦА-ГН необходимо назначение циклофосфамида внутрь (2 мг/кг/сут) или внутривенно (15 мг/кг, но не >1 г, с интервалом 2 нед первые 3 инфузии, а затем каждые 3 нед) в сочетании с кортикостероидами в высоких дозах (0,5-1 мг/кг внутрь ± пульс терапия в дозе до 1000 мг в течение 1-3 дней) (1A).
Комментарии.
Лечение циклофосфамидом продолжают в течение 3-12 мес.
Рекомендация 9. У пациентов с менее тяжелыми формами заболевания или имеющих противопоказания к циклофосфамиду следует назначать ритуксимаб и кортикостероиды в качестве альтернативного режима инициальной терапии (1B).
Комментарии.
-
Терапия ритуксимабом при АНЦА-ГН сопоставима по эффективности с циклофосфамидом и не различается по частоте побочных эффектов.
-
Поскольку сравнительные исследования различных режимов дозирования ритуксимаба не проводились, могут быть использованы оба часто используемых режима:
-
Для снижения риска инфузионных реакций введение ритуксимаба следует проводить на фоне премедикации метилпреднизолоном в/в по 250-500 мг и антигистаминными препаратами [хлоропирамин (Супрастин♠) по 20 мг в/м].
Рекомендация 10. Пациентам, нуждающимся в лечении диализом, и пациентам с быстро нарастающим СКр показано дополнительное к лечению кортикостероидами и циклофосфамидом проведение плазмафереза (1С).
Рекомендация 11. Дополнительное проведение плазмафереза рекомендовано пациентам с диффузными легочными кровотечениями (2С) и пациентам с перекрестным синдромом (сочетанием АНЦА-васкулита и анти-ГБМ ГН, в соответствии с предложенными критериями и режимами для анти-ГБМ ГН) (2D).
Рекомендация 12. У пациентов, остающихся диализ-зависимыми и не имеющих внепочечных проявлений заболевания, терапию циклофосфамидом следует отменить через 3 мес (2С).
6.2.4. Комбинированный быстропрогрессирующий гломерулонефрит (сочетание анти-БМК-нефрита и гломерулонефрита, ассоциированного с антинейтрофильными цитоплазматическими антителами) (тип IV по R. Glassock, 1997)
Рекомендация 13. При IV типе БПГН лечение нужно проводить как при анти-БМК-ГН (1В).
Комментарий. Поскольку прогноз течения анти-ГБМ хуже, чем при АНЦА-ГН, при сочетании этих типов лечение должно быть более агрессивным, чем при АНЦА-ГН.
6.2.5. Идиопатический (анти-БМК- и АНЦА-негативный гломерулонефрит) (тип V по R. Glassock, 1997)
Рекомендация 14. Больных с V типом БПГН следует лечить по протоколу, применяемому при III (АНЦА-ассоциированном ГН) типе БПГН.
Комментарий. Хотя при идиопатическом БПГН необходимость добавления цитостатиков (циклофосфамид в «пульсах» или внутрь) не доказана, применение циклофосфамида представляется оправданным.
6.3. ДАЛЬНЕЙШЕЕ ВЕДЕНИЕ БОЛЬНЫХ С БЫСТРОПРОГРЕССИРУЮЩИМ ГЛОМЕРУЛОНЕФРИТОМ
Рекомендация 15. Пациентам, достигшим ремиссии, следует проводить поддерживающую терапию в менее агрессивных режимах (1B).
Рекомендация 16. Пациентам, у которых сохраняется полная ремиссия, поддерживающую терапию следует проводить в течение не менее 18 мес (2D).
Рекомендация 17. Пациентам, остающимся диализ-зависимыми и не имеющим внепочечных проявлений заболевания, поддерживающую терапию проводить не следует (1С).
Рекомендация 18. Режим поддерживающей терапии определяет основное заболевание, в рамках которого развился БПГН (1B).
Рекомендация 19. Для лечения АГ необходимо применять антигипертензивные препараты различных групп в разных комбинациях с обязательным использованием ИАПФ или БРА с антипротеинурической и нефропротективной целями.
Рекомендация 20. Пациентам с ПУ >500 мг/с (белок/креатинин мочи >50 мг/моль) назначение ИАПФ или БРА строго обязательно.
7. Прогноз
При определении прогноза необходимо принимать во внимание клинические, лабораторные и морфологические факторы (2С).
Без лечения развитие ТПН при БПГН в течение первого года достигает 100%.
-
В результате активной иммуносупрессивной терапии ремиссия достигается у 80% пациентов.
-
При системных заболеваниях прогноз может определяться также поражением других органов (сердечно-сосудистая система, легкие).
-
Прогноз больных БПГН в первую очередь определяется тяжестью (распространенностью) поражения - количеством клубочков, имеющих полулуния:
-
при обширном поражении (полулуния в 50% клубочков и более) БПГН редко подвергается спонтанной ремиссии и при отсутствии специальной терапии почечная выживаемость не превышает 6-12 мес;
-
при небольшой степени поражения (30% клубочков и менее), особенно если полулуния наслаиваются на ранее существовавший ГН (например, IgA-нефрит, постстрептококковый), нарушенная функция почек может спонтанно восстанавливаться, иногда даже до исходного уровня;
-
при умеренном поражении (30-50% клубочков) потеря почечной функции происходит медленнее, но без лечения все равно развивается ТПН, поэтому иммуносупрессивная терапия показана всем больным БПГН, если только клинические и морфологические прогностические факторы не говорят о необратимости процесса даже при агрессивном лечении и если она не сопряжена с высоким риском осложнений.
-
Выживаемость почечного трансплантата у больных БПГН в течение 1 года достигает 90%.
8. Скрининг
Скрининг БПГН не проводят.
СПИСОК ЛИТЕРАТУРЫ
-
Захарова Е.В. Совместные рекомендации Европейской лиги по борьбе с ревматизмом и Европейской почечной ассоциации - Европейской ассоциации диализа и трансплантации по ведению волчаночного нефрита у взрослых и детей // Нефрология и диализ. 2012. Т. 14, № 4. С. 206-222.
-
Козловская Н.Л., Моисеев С.В., Новиков П.И. Лечение волчаночного нефрита: новые рекомендации EULAR/ERA-EDTA // Клин. фармакол. и тер. 2013. Т. 22, № 1. С. 62-68.
-
Лечение гломерулонефрита, обусловленного антителами к гломерулярной базальной мембране. Клинические практические рекомендации KDIGO по лечению гломерулонефритов. KDIGO Clinical Practice Guideline for Glomerulonephritis Kidney International supplements Volume 2. Issue 2. June 2012 // Нефрология и диализ. 2014. Прил. С. 136-140.
-
Мухин Н.А., Шилов Е.М. Быстропрогрессирующий гломерулонефрит // Нефрология : национальное руководство : краткое издание. М. : ГЭОТАР-Медиа, 2014. С. 210-215.
-
Пауцииммунный (малоиммунный) фокальный и сегментарный некротизирующий гломерулонефрит. Клинические практические рекомендации KDIGO по лечению гломерулонефритов. KDIGO Clinical Practice Guideline for Glomerulonephritis Kidney International supplements. Volume 2. Issue 2. June 2012 // Нефрология и диализ. 2014. Прил. С. 127-136.
-
Шилов Е.М. Экстракапиллярный (быстропрогрессирующий) гломерулонефрит // Рациональная фармакотерапия в нефрологии : руководство для практических врачей / под ред. Н.А. Мухина. М. : Литтерра, 2006. С. 242-246.
-
Adler S., Bruns F.J., Fraley D.S. et al. Rapid progressive glomerulonephritis: relapse after prolonged remission // Arch. Intern. Med. 1981. Vol. 141. C. 852-854.
-
Berden A.E., Ferrario F., Hagen E.C. et al. Histopathologic classification of ANCA-associated glomerulonephritis // J. Am. Soc. Nephrol. 2010 Oct. Vol. 21, N 10. P. 1628-1636.
-
Chen M., Kallenberg C.G. New advances in the pathogenesis of ANCA-as-sociated vasculitides // Clin. Exp. Rheumatol. 2009 Jan-Feb. Vol. 27, N 1. Suppl. 52. P. S108-S114.
-
Choy B.Y., Chan T.M., Lai K.N. Recurrent glomerulonephritis after kidney transplantation // Am. J. Transplant. 2006. Vol. 6. P. 2535-2542.
-
Cui Z., Zhao M.H., Xin G. et al. Characteristics and prognosis of Chinese patients with anti-glomerular basement membrane disease // Nephron. Clin. Pract. 2005. Vol. 99. P. c49-c55.
-
Guillevin L. Treatment of severe and/or refractory ANCA-associated vasculitis // Curr. Rheumatol. Rep. 2014 Aug. Vol. 16, N 8. P. 430.
-
Hind C.R., Bowman C., Winearls C.G. et al. Recurrence of circulating an-ti-glomerular basement membrane antibody three years after immunosuppressive treatment and plasma exchange // Clin. Nephrol. 1984. Vol. 21. P. 244-246.
-
Hirayama K., Yamagata K., Kobayashi M. et al. Anti-glomerular basement membrane antibody disease in Japan: part of the nationwide rapidly progressive glomerulonephritis survey in Japan // Clin. Exp. Nephrol. 2008. Vol. 12. P. 339-347.
-
Jindal K.K. Management of idiopathic crescentic and diffuse proliferative glomerulonephritis: evidence-based recommendations // Kidney Int. Suppl. 1999. Vol. 70. P. S33-S40.
-
Johnson J.P., Moore Jr J., Austin III H.A. et al. Therapy of anti-glomerular basement membrane antibody disease: analysis of prognostic significance of clinical, pathologic and treatment factors // Medicine (Baltimore). 1985. Vol. 64. P. 219-227.
ОСТРЫЙ ПОСТСТРЕПТОКОККОВЫЙ ГЛОМЕРУЛОНЕФРИТ
КОДИРОВАНИЕ ПО МЕЖДУНАРОДНОЙ КЛАССИФИКАЦИИ БОЛЕЗНЕЙ 10-ГО ПЕРЕСМОТРА
Класс XIV: Болезни мочеполовой системы.
Блок N00-N08: Гломерулярные болезни.
N00.4. Острый нефритический синдром - диффузный эндокапиллярный пролиферативный ГН.
N01.4. Быстропрогрессирующий нефритический синдром - диффузный эндокапиллярный й пролиферативный ГН.
N02.4. Рецидивирующая и устойчивая гематурия - диффузный эндокапиллярный пролиферативный ГН.
N03.4. Хронический нефритический синдром - диффузный эндокапиллярный пролиферативный ГН.
N04.4. НС - диффузный эндокапиллярный пролиферативный ГН.
N05.4. Нефритический синдром неуточненный - диффузный эндокапиллярный пролиферативный ГН.
N06.4. Изолированная ПУ с уточненным морфологическим поражением - диффузный эндокапиллярный пролиферативный ГН.
Введение
Острый постстрептококковый ГН (ОПСГН) - одна из форм острого постинфекционного ГН - представляет собой иммунокомплексное, обусловленное перенесенной стрептококковой инфекцией заболевание, характеризующееся разнообразными клиническими проявлениями в сочетании с морфологической картиной острого диффузного пролиферативного ГН.
ЭПИДЕМИОЛОГИЯ
ОПСГН остается наиболее частой причиной острого ГН у детей. По разным оценкам, ежегодная заболеваемость ОПСГН во всем мире составляет 470000 случаев, из них 97% приходится на регионы с низким социально-экономическим статусом, в них уровень заболеваемости колеблется от 9,5 до 28,5 случаев на 100000 населения [8, 11, 20]. В последние десятилетия в развитых странах заболеваемость ОПСГН значительно снизилась.
Пик заболеваемости приходится на возраст от 5 до 12 лет, у детей младше 2 лет ОПСГН наблюдается менее чем в 5% случаев. Во взрослой популяции риск развития ОПСГН повышен у лиц старше 60 лет [6, 9].
ОПСГН встречается в виде спорадических случаев или эпидемических вспышек (реже) после инфекций, вызванных нефритогенными штаммами стрептококка группы А. Риск заболевания зависит от локализации инфекции. В частности, частота развития ОПСГН после фарингита составляет 5-10%, а после кожной инфекции - 25% [3, 24].
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Причиной ОПСГН являются стрептококки группы А, главным образом нефритогенные М-штаммы: 1, 4, 12 (вызывают ОПСГН после фарингитов) и 2, 49, 55, 57, 60 (вызывают ОПСГН после кожных инфекций).
В настоящее время в качестве наиболее вероятного патогенетического механизма ОПСГН рассматривают отложение антигенов нефритогенных штаммов стрептококков в клубочках почек и связывание их с аутоантителами с образованием ИК in situ и активацией комплемента [19].
Природа стрептококковых антигенов до конца не установлена [5, 20, 27]. Наиболее активно обсуждается роль рецептора плазмина, ассоциированного с развитием нефрита (NAP1r) (гликолитического фермента с дегидрогеназной активностью), и стрептококкового пирогенного экзотоксина В (катионной цистеиновой протеиназы). Нефритогенный потенциал названных стрептококковых белков обусловлен их плазминсвязывающими свойствами, способностью индуцировать синтез молекул адгезии и ряда цитокинов, а также возможностью напрямую активировать систему комплемента по альтернативному пути (характерная особенность ОПСГН) [20].
МОРФОЛОГИЯ
При световой микроскопии выявляют диффузный пролиферативный ГН с преимущественно эндокапиллярной пролифераций и большим количеством нейтрофилов. Окраска трихромом позволяет в некоторых случаях обнаружить субэпителиальные отложения в виде «горбов».
При иммунофлюоресцентном исследовании в мезангии и стенках капилляров клубочков выявляют депозиты иммуноглобулина класса G (IgG) и С3-компонента комплемента диффузного гранулярного характера. Могут присутствовать IgМ, IgА, фибрин и другие компоненты комплемента.
При электронной микроскопии характерны субэпителиальные плотные депозиты в виде «горбов», которые, как и субэндотелиальные депозиты, представляют собой ИК и соответствуют обнаруживаемым при иммунофлюоресцентном исследовании отложениям IgG и С3.
КЛИНИЧЕСКАЯ КАРТИНА
Клиническая картина ОПСГН разнообразна: проявления варьируют от бессимптомной микрогематурии до развернутого остронефритического синдрома, характеризующегося развитием макрогематурии, отеков, АГ, ПУ (от минимальной до нефротического уровня), нарушением функции почек (в том числе быстропрогрессирующим) [15, 22, 23].
Характерно указание на предшествующую стрептококковую инфекцию. Длительность латентного периода ОПСГН зависит от локализации инфекции и составляет 1-3 нед после фарингита и 3-6 нед - после кожной инфекции [17].
При ОПСГН наблюдаются следующие клинические и лабораторные проявления.
-
Отеки - основная жалоба большинства пациентов. Генерализованные отеки наблюдаются в основном у детей, для взрослых характерны отеки на лице и лодыжках. Основные причины отеков - снижение фильтрации в результате повреждения клубочков, задержка натрия. Примерно у 5-10% больных развивается отек паренхимы почек, проявляющийся тупыми болями в пояснице, возможна визуализация отечной паренхимы почек при УЗИ.
-
Уменьшение объема выделяемой мочи также связано со снижением клубочковой фильтрации, задержкой натрия и жидкости. При типичном течении ОПСГН олигурия преходящая, объем мочи увеличивается через 4-7 дней с последующим быстрым исчезновением отеков и нормализацией АД.
-
АГ развивается у 50-90% больных. Выраженность АГ варьирует от мягкой до тяжелой, основная причина ее развития - увеличение объема циркулирующей крови, связанное с задержкой натрия и жидкости, а также повышение сердечного выброса и периферического сосудистого сопротивления. Гипертоническая энцефалопатия, застойная сердечная недостаточность - осложнения тяжелой АГ, требующие неотложной терапии.
-
Гематурия - обязательный симптом ОПСГН. У 30-50% больных наблюдается макрогематурия, у остальных - микрогематурия, которая может быть единственным проявлением заболевания и сохраняться в течение многих месяцев после острого периода. В свежесобранных образцах мочи обнаруживают эритроцитарные цилиндры, а при фазово-контрастной микроскопии осадка мочи выявляется более 70% дисморфных эритроцитов, указывающее на клубочковое происхождение гематурии.
-
ПУ может быть различной степени выраженности, однако, ПУ нефротического уровня выявляется редко, преимущественно у взрослых.
-
Лейкоцитурия обнаруживается примерно у 50% больных, как правило, при стерильных посевах мочи, обусловлена преимущественно лимфоцитурией, реже в сочетании с нейтрофилурией, держится недолго - 1-2 нед.
-
Цилиндрурия - обнаруживают эритроцитарные, гранулярные, и лейкоцитарные цилиндры.
-
Нарушение функции почек (повышение уровня креатинина в крови и/или снижение СКФ) в начале заболевания обнаруживают у 1/4 больных, в редких случаях наблюдается быстропрогрессирующее снижение функции почек, требующее проведения диализной терапии.
-
Повышение уровня антистрептококковых АТ. Результаты стрептозимового теста, позволяющего выявить АТ к внеклеточным продуктам стрептококка [АСЛ-О, антистрептогиалуронидазу, антистрептокиназу, антиникотинамидадениндинуклеотидазу (анти-НАД) и анти-ДНКазу В], оказываются положительными более чем в 95% случаев у больных с фарингитом и примерно в 80% случаев у пациентов с кожной инфекцией [4, 10, 12]. Эти АТ можно исследовать и отдельно: после фарингита наиболее часто повышаются уровни АСЛ-О, анти-ДНКазы В, анти-НАД и антистрептогиалуронидазы, а после инфекции кожи - только анти-ДНКазы В и антистрептогиалуронидазы.
-
Снижение уровня комплемента С3 и/или СН50 (общей гемолитической активности) наблюдается у 90% пациентов с ОПСГН в первые 2 нед заболевания [7, 14]. У некоторых больных снижаются также уровни С4 и С2, что свидетельствует об активации комплемента как по классическому, так и по альтернативному пути [6].
-
Положительные результаты посевов на стрептококк группы А обнаруживаются только у 25% больных с инфекцией носоглотки или кожи, поскольку ОПСГН возникает через несколько недель после острой стрептококковой инфекции.
Диагностика
Рекомендация 1. Диагноз ОПСГН устанавливают при выявлении клинико-лабораторных признаков острого ГН, развившихся через 1-6 нед после перенесенной инфекции, вызванной β-гемолитическим стрептококком группы А (НГ).
Диагноз ОПСГН устанавливают на основании характерных данных анамнеза, клинических признаков и результатов лабораторных исследований; решающее значение имеют следующие критерии:
Рекомендация 2. Отрицательные результаты исследования на антистрептококковые АТ у пациентов, ранее получавших антибактериальные препараты, не должны исключать диагноз перенесенной стрептококковой инфекции (НГ).
Если исследуют только уровень АСЛ-О, результат может оказаться ложно отрицательным или заниженным у пациентов с кожной инфекцией, поскольку в этом случае повышаются преимущественно титры анти-ДНКазы В и антистрептогиалуронидазы. Информативность исследования снижается при раннем начале антибактериальной терапии, подавляющей АТ-ответ.
Рекомендация 3. Мы предлагаем не проводить рутинную биопсию почки при выявлении типичных проявлений ОПСГН в сочетании с положительными серологическими тестами или посевами на β-гемолитический стрептококк и при быстрой положительной динамике клинической картины ОПСГН.
При сомнительном диагнозе ОПГН (атипичное его течение, длительное отсутствие обратного развития клинической картины и т.д.) рекомендуется проведение пункционной биопсии почки с целью уточнения морфологического варианта нефрита, оценки прогноза и определения тактики лечения (НГ).
У большинства больных с характерной клинической картиной и подтвержденной стрептококковой инфекцией биопсия почки не проводится, поскольку улучшение наступает уже в течение 1-2 нед после начала заболевания.
Биопсию почки, как правило, проводят при нетипичном течении ОПСГН для исключения других возможных заболеваний, а также при позднем начале болезни без четкой связи с недавно перенесенной стрептококковой инфекции. Показаниями к биопсии почек служат:
Дифференциальная диагностика
При типичных клинико-лабораторных проявлениях и подтверждении перенесенной стрептококковой инфекции диагноз ОПСГН в большинстве случаев не вызывает сомнений. Однако при отсутствии положительной динамики, сохранении гематурии и/или АГ более 4-6 нед, отсутствии документального подтверждения предшествующей стрептококковой инфекции необходимо исключать другие формы ГН.
Мембранопролиферативный гломерулонефрит. Клинические проявления на ранней стадии мембранопролиферативного гломерулонефрита могут быть неотличимы от проявлений ОПСГН. Мембранопролиферативный гломерулонефрит, как правило, проявляется гематурией, АГ, ПУ и гипокомплементемией, которые у некоторых пациентов возникают после острого респираторного заболевания. Однако при мембрано-пролиферативном гломерулонефрите изменения в анализах мочи и сниженный уровень комплемента сохраняются более 4-6 нед, кроме того, возможно дальнейшее повышение уровня креатинина в крови, что не характерно для ОПСГН, при котором наблюдается обратное развитие клинических проявлений и нормализация уровней С3 и СН50 в течение 1-2 нед.
IgA-нефропатия. Клинические проявления часто возникают после перенесенной инфекции верхних дыхательных путей, как и при ОПСГН. Для IgA-нефропатии, в отличие от ОПСГН, характерны более короткий интервал между острым респираторным заболеванием и появлением гематурии (менее 5 дней), наличие эпизодов макрогематурии в анамнезе, а также повышение уровня IgA сыворотки крови.
Вторичные ГН в рамках СКВ или геморрагического васкулита имеют сходные с ОПСГН проявления. Дифференцировать их позволяют наличие системных проявлений и характерные серологические тесты. В частности для геморрагического васкулита не характерна гипокомплементемия, а при СКВ наблюдаются снижение и С3, и С4, положительные АТ к ДНК, антинуклеарному фактору и др.
Другие постинфекционные ГН (ассоциированный с HBV, HCV, ГН при бактериальном эндокардите и др.) также необходимо включать в круг дифференциальной диагностики, поскольку их клинические проявления могут быть сходными с ОПСГН, но отсутствует подтверждение предшествующей стрептококковой инфекции.
Лечение
В зависимости от особенностей клиники лечение ОПСГН включает этиотропную, патогенетическую, симптоматическую терапию и лечение осложнений.
ПОКАЗАНИЯ К ГОСПИТАЛИЗАЦИИ
Рекомендация 4. Предлагается адекватное лечение инфекционного заболевания, ставшего причиной ОПСГН, и стандартные подходы к лечению проявлений ОПСГН (2D).
ЛЕЧЕНИЕ ИНФЕКЦИОННОГО ЗАБОЛЕВАНИЯ
Всем пациентам с документально подтвержденной стрептококковой инфекцией (положительные результаты посевов с кожи, зева и выявление высоких титров антистрептококковых АТ в крови) рекомендуется проводить антибактериальную терапию. Назначение антибиотиков также показано больным с клинической триадой (лихорадка, увеличение небных миндалин и шейных лимфоузлов). При наличии одного или двух из трех указанных симптомов бактериальные препараты назначают в случае получения положительных результатов бактериологического исследования.
Для предупреждения инфицирования нефритогенными штаммами стрептококка А в период эпидемии оправдано профилактическое назначение антибиотиков лицам, тесно контактирующим с пациентом.
Антибактериальную терапию проводят с учетом чувствительности возбудителя. Наиболее часто назначают препараты пенициллинового ряда. Макролиды II и III поколений являются препаратами второй линии терапии.
У пациентов с уже выявленными признаками иммунокомплексного ГШ антибиотики не способствуют обратному развитию заболевания, антибактериальная терапия проводится с целью устранения очага инфекции.
ЛЕЧЕНИЕ ГЛОМЕРУЛОНЕФРИТА
Общие принципы лечения ГН включают немедикаментозную (соблюдение режима и диеты), симптоматическую и патогенетическую терапию в соответствии с особенностями клинического течения ОПСГН и развивающимися осложнениями.
-
Режим - постельный при выраженных отеках, макрогематурии, умеренной/тяжелой АГ, сердечной недостаточности (обычно в первые 3-4 нед). При улучшении состояния режим постепенно расширяют.
-
-
С ограничением потребления соли (до 1-2 г/сут) и жидкости в острый период болезни, особенно при быстром нарастании отеков, олигурии и АГ. Объем жидкости рассчитывают исходя из диуреза за предыдущий день с учетом внепочечных потерь, прием жидкости не должен превышать диуреза более чем на 200 мл.
-
С ограничением белка до 0,5 г/кг/сут при снижении функции почек менее 60 мл/мин (до нормализации СКФ и уровня креатинина в крови, но не длительнее 2-4 нед).
-
Симптоматическая терапия направлена на:
При отеках и АГ патогенетически обосновано назначение мочегонных средств, препаратами выбора являются петлевые диуретики, которые, увеличивая натрийурез и фильтрацию, уменьшают задержку жидкости и выраженность отеков и АГ. Однако умеренные отеки и гипертония сразу не требуют назначения мочегонных, вначале оправданы ограничение натрия и жидкости.
Терапия диуретиками показана при:
Для лечения повышенного АД, наряду с мочегонными, предпочтительны блокаторы БКК; ИАПФ или БРА следует назначать с осторожностью. Применение последних возможно при сохранной функции почек и отсутствии гиперкалиемии.
При выраженной гиперкоагуляции и активации внутрисосудистого свертывания (в том числе при НС) возможно назначение антикоагулянтов.
Иммуносупрессивная терапия
Рекомендация 5. При высокоактивном течении ОПСГН (НС, быстропрогрессирующая ПН, наличие более 30% полулуний в биоптате) предлагается терапия глюкокортикостероидами.
При быстропрогрессирующем течении ОПСГН и/или выявлении более 30% полулуний в биоптате почки предлагается проведение пульс-терапии метилпреднизолоном в соответствии с подходами к лечению быстропрогрессирующего и полулунного ГН.
При сохраняющемся более 2 нед НС, стабильно повышенном уровне креатинина (без тенденции к дальнейшему нарастанию и нормализации) и при невозможности проведения биопсии почки рекомендуется терапия преднизолоном внутрь в дозе 1 мг/кг/сут (2/3 дозы в утренний прием после еды, 1/3 дозы в дневной прием после еды) в течение 1-2 мес.
В отдельных наблюдениях подобных групп пациентов с ОПСГН применение глюкокортикостероидов способствовало положительной динамике клинической картины, однако РКИ, доказывающие эффективность такого подхода при ОПСГН, отсутствуют [18, 21].
Заместительная почечная терапия
Некоторым пациентам с нарушением функции почек может потребоваться проведение ЗПТ (см. Рекомендации по лечению острого почечного повреждения).
Течение
В большинстве случаев (особенно у детей) наблюдается быстрое разрешение клинических проявлений: диурез восстанавливается в течение первой недели, уровень креатинина возвращается к исходному значению через 3-4 нед [15].
Сроки нормализации анализов мочи различны. Гематурия, как правило, исчезает через 3-6 мес. ПУ снижается медленнее; у 15% следовая ПУ может сохраняться более года.
В тяжелых случаях ПУ нефротического уровня может персистировать до 6 мес и более, даже после исчезновения гематурии), что требует начала терапии стероидами.
Параллельно эволюции клинических проявлений наблюдается положительная динамика гистологических изменений, в частности существенно снижается число клеток воспаления и иммунных депозитов [25]. Более медленное по сравнению с темпами исчезновения гематурии и восстановлением функции почек снижение ПУ объясняется более длительным сохранением иммунных депозитов, особенно субэпителиальной локализации. В целом степень ПУ коррелирует с числом субэпителиальных депозитов.
Рецидивы ОПСГН наблюдаются крайне редко и могут быть обусловлены длительным персистированием АТ к нефритогенным антигенам стрептококка.
Прогноз
Ближайший прогноз при ОПСГН, в целом, благоприятный [5, 16]. У детей и взрослых угрожающие жизни осложнения отечного синдрома и эклампсия встречаются редко. У пациентов пожилого возраста в остром периоде ОПСГН значительно чаще, чем в общей популяции, наблюдаются одышка, застойные явления в легких, острая сердечная недостаточность, ОПН и смерть [16, 26].
Отдаленный прогноз в целом благоприятный: частота развития ТПН составляет менее 1%. У пожилых пациентов с персистирующей ПУ прогноз хуже [20].
Факторами неблагоприятного прогноза служат:
СПИСОК ЛИТЕРАТУРЫ
-
Длин В.В., Приходина Л.С. Острый постстрептококковый гломерулонефрит // Педиатрия : национальное руководство / под ред. А.А. Баранова. М. : ГЭОТАР-Медиа, 2009. 1024 с.
-
Мухин Н.А., Козловская Л.В., Шилов Е.М. и др. Рациональная фармакотерапия в нефрологии : руководство для практикующих врачей. М. : Литтерра, 2006. 896 с.
-
Anthony B.F., Kaplan E.L., Wannamaker L.W. et al. Attack rates of acute nephritis after type 49 streptococcal infection of the skin and of the respiratory tract // J. Clin. Invest. 1969. Vol. 48. P. 1697-1704.
-
Ayoub E.M., Wannamaker L.W. Streptococcal antibody titers in Sydenham’s chorea // Pediatrics. 1966. Vol. 38. P. 946-956.
-
Batsford S.R., Mezzano S., Mihatsch M. et al. Is the nephritogenic antigen in post-streptococcal glomerulonephritis pyrogenic exotoxin B (SPE B) or GAP-DH? // Kidney Int. 2005. Vol. 68. P. 1120-1129.
-
Blyth C.C., Robertson P.W., Rosenberg A.R. Post-streptococcal glomeru-lonephritis in Sydney: a 16-year retrospective review // J. Paediatr. Child Health. 2007. Vol. 43. P. 446-450.
-
Cameron J.S., Vick R.M., Ogg C.S. et al. Plasma C3 and C4 concentrations in management of glomerulonephritis // Br. Med. J. 1973. Vol. 3. P. 668-672.
-
Carapetis J.R., Steer A.C., Mulholland E.K., Weber M. The global burden of group A streptococcal diseases // Lancet Infect. Dis. 2005. Vol. 5. P. 685-694.
-
Coppo R., Gianoglio B., Porcellini M.G., Maringhini S. Frequency of renal diseases and clinical indications for renal biopsy in children (report of the Italian National Registry of Renal Biopsies in Children). Group of Renal Immunopathol-ogy of the Italian Society of Pediatric Nephrology and Group of Renal Immuno-pathology of the Italian Society of Nephrology // Nephrol. Dial. Transplant. 1998. Vol. 13. P. 293-297.
-
Eison T.M., Ault B.H., Jones D.P. et al. Post-streptococcal acute glomerulonephritis in children: clinical features and pathogenesis // Pediatr. Nephrol. 2011. Vol. 26. P. 165-180.
-
Jackson S.J., Steer A.C., Campbell H. Systematic review: Estimation of global burden of nonsuppurative sequelae of upper respiratory tract infection: rheumatic fever and post-streptoccocal glomerulonephritis // Trop. Med. Int. Health. 2011. Vol. 16, N 1. P. 2-11.
-
Kaplan E.L., Anthony B.F., Chapman S.S. et al. The influence of the site of infection on the immune response to group A streptococci // J. Clin. Invest. 1970. Vol. 49. P. 1405-1414.
-
Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis WorkGroup. KDIGO Clinical Practice Guideline for Glomerulonephritis // Kidney Int. Suppl. 2012. Vol. 2. P. 139-274.
-
Lewis E.J., Carpenter C.B., Schur P.H. Serum complement component levels in human glomerulonephritis // Ann. Intern. Med. 1971. Vol. 75. P. 555-560.
-
Lewy J.E., Salinas-Madrigal L., Herdson P.B. et al. Clinico-pathologic correlations in acute poststreptococcal glomerulonephritis. A correlation between renal functions, morphologic damage and clinical course of 46 children with acute poststreptococcal glomerulonephritis // Medicine (Baltimore). 1971. Vol. 50. P. 453-501.
-
Melby P.C., Musick W.D., Luger A.M., Khanna R. Poststreptococcal glo-merulonephritis in the elderly. Report of a case and review of the literature // Am. J. Nephrol. 1987. Vol. 7, N 3. P. 235-240.
-
Nissenson A.R., Baraff L.J., Fine R.N., Knutson D.W. Poststreptococcal acute glomerulonephritis: fact and controversy // Ann. Intern. Med. 1979. Vol. 91. P. 76-86.
-
Raff A., Hebert T., Pullman J., Coco M. Crescentic post-streptococcal glo-merulonephritis with nephrotic syndrome in the adult: is aggressive therapy warranted? // Clin. Nephrol. 2005. Vol. 63. P. 375-380.
-
Rodriguez-Iturbe B., Batsford S. Pathogenesis of poststreptococcal glomerulonephritis a century after Clemens von Pirquet // Kidney Int. 2007. Vol. 71. P. 1094-1104.
-
Rodriguez-Iturbe B., Musser J.M. The current state of poststreptococcal glomerulonephritis // J. Am. Soc. Nephrol. 2008. Vol. 19. P. 1855-1864.
-
Roy S. 3rd, Murphy W.M., Arant B.S. Jr. Poststreptococcal crescenteric glomerulonephritis in children: comparison of quintuple therapy versus supportive care // J. Pediatr. 1981. Vol. 98. P. 403-410.
-
Sagel I., Treser G., Ty A. et al. Occurrence and nature of glomerular lesions after group A streptococci infections in children // Ann. Intern. Med. 1973. Vol. 79. P. 492-499.
-
Sanjad S., Tolaymat A., Whitworth J., Levin S. Acute glomerulonephritis in children: a review of 153 cases // South. Med. J. 1977. Vol. 70. P. 1202-1206.
-
Stetson C.A., Rammelkamp C.H.Jr, Krause R.M. et al. Epidemic acute nephritis: studies on etiology, natural history and prevention // Medicine (Baltimore). 1955. Vol. 34. P. 431-450.
-
Tornroth T. The fate of subepithelial deposits in acute poststreptococcal glomerulonephritis // Lab. Invest. 1976. Vol. 35. P. 461-474.
-
Washio M., Oh Y., Okuda S. et al. Clinicopathological study of poststreptococcal glomerulonephritis in the elderly // Clin. Nephrol. 1994 May. Vol. 41, N 5. P. 265-270.
-
Yoshizawa N., Yamakami K., Fujino M. et al. Nephritis-associated plasmin receptor and acute poststreptococcal glomerulonephritis: characterization of the antigen and associated immune response // J. Am. Soc. Nephrol. 2004. Vol. 15. P. 1785-1793.
ОТДЕЛЬНЫЕ ФОРМЫ ПОСТИНФЕКЦИОННОГО ГЛОМЕРУЛОНЕФРИТА: ГЛОМЕРУЛОНЕФРИТ ПРИ ИНФЕКЦИОННОМ ЭНДОКАРДИТЕ И ШУНТ-НЕФРИТ
КОДИРОВАНИЕ ПО МЕЖДУНАРОДНОЙ КЛАССИФИКАЦИИ БОЛЕЗНЕЙ 10-ГО ПЕРЕСМОТРА. КЛАСС XIV: БОЛЕЗНИ МОЧЕПОЛОВОЙ СИСТЕМЫ. БЛОК N00-N08: ГЛОМЕРУЛЯРНЫЕ БОЛЕЗНИ
N00. |
Острый нефритический синдром |
|
0 |
Незначительные гломерулярные нарушения |
|
1 |
Очаговые и сегментарные гломерулярные повреждения |
|
3 |
Диффузный мезангиальный пролиферативный ГН |
|
4 |
Диффузный эндокапиллярный пролиферативный ГН |
|
5 |
Диффузный мезангиокапиллярный ГН |
|
N01. |
Быстропрогрессирующий нефритический синдром |
|
0 |
Незначительные гломерулярные нарушения |
|
1 |
Очаговые и сегментарные гломерулярные повреждения |
|
3 |
Диффузный мезангиальный пролиферативный ГН |
|
4 |
Диффузный эндокапиллярный пролиферативный ГН |
|
5 |
Диффузный мезангиокапиллярный ГН |
|
N02. |
Рецидивирующая и устойчивая гематурия |
|
0 |
Незначительные гломерулярные нарушения |
|
1 |
Очаговые и сегментарные гломерулярные повреждения |
|
3 |
Диффузный мезангиальный пролиферативный ГН |
|
4 |
Диффузный эндокапиллярный пролиферативный ГН |
|
5 |
Диффузный мезангиокапиллярный ГН |
|
N03. |
Хронический нефритический синдром |
|
0 |
Незначительные гломерулярные нарушения |
|
1 |
Очаговые и сегментарные гломерулярные повреждения |
|
3 |
Диффузный мезангиальный пролиферативный ГН |
|
4 |
Диффузный эндокапиллярный пролиферативный ГН |
|
5 |
Диффузный мезангиокапиллярный ГН |
|
N04. |
НС |
|
0 |
Незначительные гломерулярные нарушения |
|
1 |
Очаговые и сегментарные гломерулярные повреждения |
|
3 |
Диффузный мезангиальный пролиферативный ГН |
|
4 |
Диффузный эндокапиллярный пролиферативный ГН |
|
5 |
Диффузный мезангиокапиллярный ГН |
|
N05. |
Нефритический синдром неуточненный |
|
0 |
Незначительные гломерулярные нарушения |
|
1 |
Очаговые и сегментарные гломерулярные повреждения |
|
3 |
Диффузный мезангиальный пролиферативный ГН |
|
4 |
Диффузный эндокапиллярный пролиферативный ГН |
|
5 |
Диффузный мезангиокапиллярный ГН |
|
N06. |
Изолированная ПУ с уточненным морфологическим поражением |
|
0 |
Незначительные гломерулярные нарушения |
|
1 |
Очаговые и сегментарные гломерулярные повреждения |
|
3 |
Диффузный мезангиальный пролиферативный ГН |
|
4 |
Диффузный эндокапиллярный пролиферативный ГН |
|
5 |
Диффузный мезангиокапиллярный ГН |
|
1. Гломерулонефрит при инфекционном эндокардите
ВВЕДЕНИЕ
Инфекционный эндокардит (ИЭ) - воспалительное поражение клапанных структур сердца и пристеночного эндокарда, обусловленное различными возбудителями, протекающее чаще всего по типу сепсиса (острого или подострого) с циркуляцией возбудителя в крови, неспецифическими системными проявлениями, тромбогеморрагическими и иммунными изменениями.
ЭПИДЕМИОЛОГИЯ
Заболеваемость ИЭ колеблется в разных странах от 3 до 10 случаев на 100000 населения в год [5, 14, 15, 30]. За последние несколько лет эпидемиология ИЭ существенно изменилась. Ранее заболевали преимущественно пациенты молодого возраста с предшествовавшим (преимущественно ревматическим) поражением клапанов [14], а в настоящее время в развитых странах возросла частота ИЭ у больных более старшего возраста (в результате диагностических и лечебных процедур), у лиц без предшествующего поражения клапанов, у пациентов с протезированными клапанами, у инъекционных наркоманов [13].
ЭТИОЛОГИЯ
Наиболее частыми возбудителями ИЭ являются стрептококки (главным образом зеленящие Streptococcus viridans) и стафилококки (преимущественно золотистый Staphylococcus aureus, эпидермальный Staphylococcus epidermidis); в последние десятилетия роль Staphylococcus aureus в этиологии ИЭ существенно возросла [20]. В развитых странах основными факторами риска развития ИЭ являются хронический ГД, наличие СД, применение внутрисосудистых устройств, внутривенное введение лекарственных препаратов [7, 9, 27]. Другими возбудителями могут быть энтерококки, пневмококки, грамотрицательные микроорганизмы, хламидии, риккетсии, бруцеллы, грибы. В 5-15% случаев ИЭ возбудителя выделить не удается.
При ИЭ у 50-80% больных выявляют поражение почек, которое может быть нескольких типов:
Примерно у 30% пациентов развивается острое повреждение почек (ОПП), обусловленное разными причинами (эмболия в почечной артерии, гемодинамические нарушения, лекарственное, иммунное повреждение). Чаще ОПП наблюдается у лиц старшего возраста или при инфицировании Staphylococcus aureus [8]. У 10% пациентов с поражением почек развивается ХПН.
ПАТОГЕНЕЗ ГЛОМЕРУЛОНЕФРИТА ПРИ ИНФЕКЦИОННОМ ЭНДОКАРДИТЕ
В основе развития ГН при ИЭ лежит классический иммунокомплексный механизм. В ответ на попадающие в кровь бактериальные антигены происходит выработка соответствующих АТ и формирование циркулирующих ИК, которые, фиксируясь в клубочках почки, вызывают активацию комплемента с последующим повреждением клубочка и развитием ГН.
МОРФОЛОГИЧЕСКАЯ КАРТИНА ГЛОМЕРУЛОНЕФРИТА ПРИ ИНФЕКЦИОННОМ ЭНДОКАРДИТЕ
Гистологически при ИЭ различают фокальный и диффузный пролиферативный ГН. Диффузный пролиферативный ГН наблюдается у 20% больных, наиболее часто характеризуется картиной МБПГН и экстракапиллярного ГН, реже - мезангиопролиферативного ГН. У некоторых пациентов могут быть обнаружены диффузные эндокапиллярные повреждения с полулуниями или без них. Возможен вариант малоиммунного ГН с полулуниями [19].
Особенностями нефрита при ИЭ является частое обнаружение васкулитов внегломерулярных сосудов и разной степени выраженности тубулоинтерстициальных изменений (лимфогистиоцитарная инфильтрация, атрофия канальцев, фиброз интерстиция).
При иммуногистохимическом исследовании выявляются гломерулярные отложения ИК (мезангиальные, субэндотелиальные, субэпителиальные), содержащие иммуноглобулины классов А, М, G и C3.
КЛИНИКО-ЛАБОРАТОРНЫЕ ИЗМЕНЕНИЯ ПРИ ГЛОМЕРУЛОНЕФРИТЕ, АССОЦИИРОВАННОМ С ИНФЕКЦИОННЫМ ЭНДОКАРДИТОМ
Клиническая картина ГН при ИЭ аналогична наблюдаемой при первичном ГН; характерны следующие клинические и лабораторные проявления [18, 24, 28].
-
Гематурия (часто макрогематурия) в сочетании с лейкоцитурией и/или эритроцитарными цилиндрами.
-
ПУ (различной степени выраженности). НС развивается в 10-30% случаев.
-
Нарушение функции почек (повышение уровня креатинина в крови и/или снижение СКФ), в том числе быстропрогрессирующее.
-
Снижение уровня комплемента (общей гемолитической активности СН50 и его фракций С3 и С4).
-
Повышение содержания циркулирующих ИК, содержащих в своем составе бактериальный антиген.
-
Возможно выявление АНЦА (редко) в сочетании с клинической картиной БПГН и наличием в биоптате почки малоиммунного ГН и полулуний.
ДИАГНОСТИКА ГЛОМЕРУЛОНЕФРИТА ПРИ ИНФЕКЦИОННОМ ЭНДОКАРДИТЕ
Диагноз ГН при ИЭ устанавливают на основании характерных данных анамнеза, клинических признаков и результатов лабораторных исследований; решающее значение имеют следующие критерии:
При современном течении ИЭ типичные симптомы заболевания (интермиттирующая лихорадка с ознобом и потом, формирование клапанного порока, тромбэмболический синдром) могут отсутствовать, а на первый план выступают неспецифические симптомы васкулита, анемия, иммунные сдвиги, поражение почек. В ряде случаев симптомы поражения почек могут предшествовать развернутой клинической картине ИЭ (так называемая нефритическая маска ИЭ).
Большинству больных ГН при ИЭ не проводится морфологическое исследование почечной ткани из-за опасности выполнения биопсии почки в условиях бактериемии или в связи с улучшением клинических проявлений ГН при адекватной антибактериальной терапии ИЭ.
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
ГН при ИЭ необходимо дифференцировать с другими вариантами поражения почек при данном заболевании.
Лекарственный острый тубулоинтерстициальный нефрит (как правило, обусловленный применением пенициллинов, цефалоспоринов или фторхинолонов) имеет сходные с ГН проявления: наблюдается гематурия (иногда с эритроцитарными цилиндрами), умеренно выраженная ПУ, нарушение функции почек. Пиурия и лейкоцитарные цилиндры более характерны для ОТИН. Важное значение имеет время манифестации поражения почек. ГН, как правило, развивается в период развернутой клинической картины ИЭ и до начала антибактериальной терапии, тогда как ОТИН в большинстве случаев развивается через 10 и более дней лечения [16, 25]. В его пользу свидетельствует также выявление эозинофилии и эозинофилурии. Рецидив лихорадки, обусловленной ОТИН, часто ошибочно расценивают как следствие неэффективности антибактериальной терапии.
Острый канальцевый некроз развивается вследствие токсического действия ряда антибиотиков или в результате гемодинамических нарушений с ишемией тубулоинтерстиция, которые могут вызывать нестероидные противовоспалительные средства или мочегонные препараты. Характеризуется клиникой ОПН (олигурия/анурия, нарастающая азотемия, снижение СКФ), изменения в анализе мочи варьируют от «пустого» осадка до появления многочисленных зернистых цилиндров грязно-коричневого цвета, клеток почечного эпителия и эпителиальных цилиндров.
Инфаркт почки наблюдается у 30-60% больных ИЭ, вызван эмболией почечных артерий (реже васкулитом почечных сосудов), как правило, развивается в период развернутой клинической картины ИЭ, но может наблюдаться и через несколько месяцев после эрадикации возбудителя. Проявляется интенсивными болями в пояснице или эпигастральной области, макрогематурией, повышением АД, тошнотой, задержкой стула. Часто сочетается с эмболиями в сосудах других органов.
Амилоидоз возникает у 3-5% больных при длительном течении ИЭ, характеризуется неуклонным нарастанием ПУ с развитием НС; особенностью является развитие ХПН при сохраняющемся НС.
ИЭ необходимо дифференцировать с асептическим тромботическим эндокардитом, который может осложнять течение опухолевого процесса, СД, АФС и протекать с признаками поражения почек. Дифференцировать их позволяют серологические тесты (выявление маркеров АФС, СКВ, онкомаркеров), положительная гемокультура [23].
ЛЕЧЕНИЕ
Рекомендация. Предлагается адекватное лечение ИЭ, ставшего причиной ГН, и стандартные подходы к лечению проявлений ГН (НГ).
Лечение инфекционного заболевания
Всем пациентам с документально подтвержденным ИЭ рекомендуется проводить его терапию в соответствии с общепринятыми современными рекомендациями (см. соответствующие руководства) [21].
Лечение должно осуществляться под контролем уровней СН50, который можно рассматривать как маркер эффективности терапии. Замедленный темп нормализации уровня комплемента указывает на персистирование инфекции и необходимость коррекции терапии.
Лечение гломерулонефрита
Общие принципы лечения ГН включают немедикаментозную (соблюдение режима и диеты), симптоматическую и патогенетическую терапию в соответствии с особенностями клинического течения острого постстрептококкового гломерулонефрита (ОПСГН) и развивающимися осложнениями.
-
Режим - постельный при выраженных отеках, макрогематурии, умеренной/тяжелой АГ, сердечной недостаточности (обычно в первые 3-4 нед). При улучшении состояния режим постепенно расширяют.
-
-
С ограничением потребления соли (до 1-2 г/сут) и жидкости в острый период болезни, особенно при быстром нарастании отеков, олигурии и АГ. Объем жидкости рассчитывают исходя из диуреза за предыдущий день с учетом внепочечных потерь, прием жидкости не должен превышать диурез более чем на 200 мл.
-
С ограничением белка до 0,5 г/кг/сут при снижении функции почек менее 60 мл/мин (до нормализации СКФ и уровня креатинина в крови, но не длительнее 2-4 нед).
-
Симптоматическая терапия направлена на:
При отеках и АГ патогенетически обосновано назначение мочегонных средств, препаратами выбора являются петлевые диуретики, которые, увеличивая натрийурез и фильтрацию, уменьшают задержку жидкости и выраженность отеков и АГ. Однако умеренные отеки и гипертония сразу не требуют назначения мочегонных, вначале оправданы ограничение натрия и жидкости.
Терапия диуретиками показана при:
Для лечения повышенного АД, наряду с мочегонными, предпочтительны блокаторы медленных кальциевых каналов (БКК); ИАПФ или БРА следует назначать с осторожностью. Применение последних возможно при сохранной функции почек и отсутствии гиперкалиемии.
При выраженной гиперкоагуляции и активации внутрисосудистого свертывания (в том числе при НС) возможно назначение антикоагулянтов.
Иммуносупрессивная терапия. При нарушении функции почек у пациентов с ГН, сохраняющимся, несмотря на адекватную антибактериальную терапию ИЭ, возможно назначение глюкокортикостероидов, однако доказательная база эффективности и безопасности такого лечения недостаточна, требуется проведение многоцентровых исследований.
ЗПТ. Некоторым пациентам с нарушением функции почек может потребоваться проведение ЗПТ (см. Рекомендации по лечению острого почечного повреждения).
ПРОГНОЗ ГЛОМЕРУЛОНЕФРИТА ПРИ ИНФЕКЦИОННОМ ЭНДОКАРДИТЕ
Ближайший прогноз ГН в целом хороший.
Тяжесть ГН в основном обусловлена длительностью персистирования инфекции до начала антибактериальной терапии. Устранение инфекции в большинстве случаев приводит к быстрому выздоровлению с полным или частичным восстановлением функции почек. Тем не менее при отсроченном начале антибактериальной терапии возможно развитие необратимых структурных и функциональных нарушений [18, 19, 24].
Шунт-нефрит
Шунт-нефрит - вариант постинфекционного ГН, который развивается у больных с инфицированными вентрикулоатриальными или вентрикуло-югулярными шунтами, установленными для устранения врожденной или приобретенной окклюзионной гидроцефалии.
Частота инфицирования шунта, по разным данным, варьирует от 10 до 30% [3, 17]. Среди больных с инфекцией шунта пациенты с ГН составляют лишь незначительную часть (от 0,7 до 2,3%) [4, 26, 29].
Первичное событие при инфицировании шунта - колонизация возбудителя в дистальной (атриальной) части шунтирующей системы, либо в момент операции по ее установке, либо, чаще, вследствие транзиторной бактериемии.
В большинстве случаев возбудителем являются стафилококки (S. epidermidis [до 75% случаев] и S. aureus), но описаны инфекции шунта, вызванные другими микроорганизмами: E. coli, Corynebacterium bovis, Corynebacterium xerosis, Pseudomonas aeruginosa, Pseudomonas cepacia, diphteroids, Listeria monocytogenes, Bacillus subtilis, Bacillus cereus, Serratia, Propionibacterium acnes, Peptococcus, Mycobacterium gordonae, Micrococcus, Moraxella nonliquefaciens, Moraxella bovis, Acinetobacter iowoffi, α-гемолитическим стрепрококком и грибами рода Fusarium [2, 6, 10, 22, 31] Отрицательные результаты посевов крови или цереброспинальной жидкости не исключают диагноз шунт-нефрита, особенно при наличии предшествующей антибактериальной терапии.
ПАТОГЕНЕЗ
Патогенез шунт-нефрита сходен с патогенезом поражения почек при ИЭ - в обоих случаях ведущим механизмом гломерулярного повреждения является отложение в клубочках ИК, содержащих бактериальный антиген.
Появление признаков инфекции в пределах нескольких месяцев после установки вентрикуло-атриального шунта свидетельствует о его контаминации непосредственно во время операции. Если признаки инфекции появляются позже, следует исключать другие пути заражения.
В частности это могут быть микроповреждения кожи или хирургические манипуляции, способствующие попаданию в кровь S. epidermidis, обладающего высокой аффинностью к гидрофобным материалам, из которых изготовлены вентрикуло-атриальные катетеры. Рост бактерий приводит к транзиторной бактериемии, высвобождению бактериальных антигенов и выработке АТ к ним, образованию циркулирующих ИК, которые затем откладываются в клубочках.
Преобладающим морфологическим вариантом шунт-нефрита является мембранопролиферативный (мезангиокапиллярный) ГН 1го типа. При иммунофлюоресцентном исследовании выявляют отложения IgM, IgG и комплемента (фракция С3) гранулярного характера, при электронной микроскопии обнаруживают электронноплотные субэндотелиальные и мезангиальные депозиты [12].
У 13-32% больных шунт-нефритом обнаруживают диффузный мезангиальный пролиферативный нефрит, у 15% - эндо- или экстракапиллярный ГН.
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ ШУНТ-НЕФРИТА
Наиболее частыми клиническими симптомами шунт-нефрита являются гематурия (обычно микрогематурия, но встречается и персистирующая макрогематурия) и ПУ (нередко - выраженная: у половины больных развивается НС). Артериальная гипертония возникает примерно в половине случаев; частота нарушения функции почек достигает 60%. В ряде случаев шунт-нефрит протекает с клиникой БПГН [12].
При серологическом исследовании у 85-90% больных обнаруживают снижение С3-фракции комплемента.
Положительные результаты посевов крови и CV: выявляют в 70-86% и 43-80% случаев соответственно.
У большинства больных интервал от момента имплантации шунта до появления первых симптомов поражения почек довольно продолжительный (в среднем составляет 4 года). Поражению почек, как правило, предшествуют эпизоды лихорадки, анемия, спленомегалия, у многих пациентов наблюдаются неврологические симптомы (головные боли, сонливость, тошнота, рвота), обусловленные внутричерепной гипертензией, развившейся вследствие нарушения функции шунта.
ДИАГНОСТИКА
Диагноз устанавливают при сочетании признаков поражения почек с клиникой сепсиса у больных с установленными вентрикуло-сосудистыми шунтами.
Дифференциальная диагностика проводится с:
ЛЕЧЕНИЕ
Рекомендация. Предлагается адекватное лечение инфекции AB или вентрикуло-югулярного шунта, ставшей причиной развития ГН, и стандартные подходы к лечению проявлений ГН (НГ).
Лечение шунт-нефрита заключается в удалении инфицированного шунта и проведении терапии в соответствии с типом выделенного возбудителя и согласно современным рекомендациям по его эрадикации.
При ранней установке диагноза и своевременном начале терапии прогноз шунт-нефрита благоприятный: полное выздоровление наблюдается в 50% случаев. Примерно у 20% больных изменения в анализах мочи сохраняются.
Тяжесть шунт-нефрита определяется главным образом временем, прошедшим с момента инфицирования до начала антибактериальной терапии. Поздняя диагностика и, как следствие, длительное персистирование инфекции и позднее начало терапии приводят к прогрессированию поражения почек и ухудшению прогноза. По разным данным, ТПН развивается у 6-19% больных, примерно с такой же частотой наблюдаются летальные осложнения [12].
СПИСОК ЛИТЕРАТУРЫ
-
Мухин Н.А., Козловская Л.В., Шилов Е.М. и др. Рациональная фармакотерапия в нефрологии : руководство для практикующих врачей. М. : Литтерра, 2006. 896 с.
-
Arze R.S., Rashid H., Morley R. et al. Shunt nephritis: report of two cases and review of the literature // Clin. Nephrol. 1983. Vol. 19. P. 48.
-
Ascho A. In vitro Testung von Hydrocephalus Ventilen. [Thesis]. Ruperto Carola University of Heidelberg, 1994. P. 305-308.
-
Bayston R., Swinden J. The aetiology and prevention of shunt nephritis // Z. Kinderchir. 1979. Vol. 28. P. 377-384.
-
Berlin J.A., Abrutyn E., Strom B.L. et al. Incidence of infective endocarditis in the Delaware Valley, 1988-1990 // Am. J. Cardiol. 1995. Vol. 76. P. 933-936.
-
Bogdanovic R., Marjanovic B., Nikolic V. et al. Shunt nephritisassociated with Moraxella bovis // Acta Paediatr. 1996. Vol. 85. P. 882-883.
-
Cabell C.H. Jr., Jollis J.G., Peterson G.E. et al. Changing patient characteristics and the effect on mortality in endocarditis // Arch. Intern. Med. 2002. Vol. 162. P. 90-94.
-
Conlon P.J., Jefferies F., Krigman H.R. et al. Predictors of prognosis and risk of acute renal failure in bacterial endocarditis // Clin. Nephrol. 1998. Vol. 49. P. 96-101.
-
Fowler V.G. Jr, Miro J.M., Hoen B. et al. Staphylococcus aureus endocarditis: a consequence of medical progress // JAMA. 2005. Vol. 293. P. 3012-3021.
-
Groeneveld A.B.J., Nommensen F.E., Mullink H. et al. Shunt nephritis associated with Propionibacterium acnes with demonstration of the antigen in the glomeruli // Nephron. 1982. Vol. 32. P. 365-369.
-
Habib G., Hoen B., Tornos P. et al. Guidelines on the prevention, diagnosis, and treatment of infective endocarditis (new version 2009): the Task Force on the Prevention, Diagnosis, and Treatment of Infective Endocarditis of the European Society of Cardiology (ESC). Endorsed by the European Society of Clinical Microbiology and Infectious Diseases (ESCMID) and the International Society of Chemotherapy (ISC) for Infection and Cancer // Eur. Heart J. 2009. Vol. 30. P. 2369-2413.
-
Haffner D., Schindera F., Aschoff A. et al. The clinical spectrum of shunt nephritis // Nephrol. Dial. Transplant. 1997. Vol. 12. P. 1143-1148.
-
Hill E.E., Herijgers P., Claus P. et al. Infective endocarditis: changing epidemiology and predictors of 6-month mortality: a prospective cohort study // Eur. Heart J. 2007. Vol. 28. P. 196-203.
-
Hoen B., Alla F., Selton-Suty C., Beguinot I. et al. Changing profile of infective endocarditis: results of a 1-year survey in France // JAMA. 2002. Vol. 288. P. 75-81.
-
Hogevik H., Olaison L., Andersson R. et al. Epidemiologic aspects of infective endocarditis in an urban population. A 5-year prospective study // Medicine (Baltimore). 1995. Vol. 74. P. 324-339.
-
Kannan S., Mattoo T.K. Diffuse crescentic glomerulonephritis in bacterial endocarditis // Pediatr. Nephrol. 2001. Vol. 16. P .423.
-
Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephri-tis WorkGroup. KDIGO Clinical Practice Guideline for Glomerulonephritis // Kidney Int. Suppl. 2012. Vol. 2. P. 139-274.
-
Koya D., Shibuya K., Kikkawa R. et al. Successful recovery of infective endocarditis-induced rapidly progressive glomerulonephritis by steroid therapy combined with antibiotics: a case report // BMC Nephrol. 2004. Vol. 5. P. 18.
-
Majumdar A., Chowdhary S., Ferreira M.A. et al. Renal pathological findings in infective endocarditis // Nephrol. Dial. Transplant. 2000. Vol. 15. P. 1782-1787.
-
Moreillon P., Que Y.A. Infective endocarditis // Lancet. 2004. Vol. 363. P. 139-149.
-
Moyssakis I., Tektonidou M.G., Vasilliou V.A. et al. Libman-Sacks endocarditis in systemic lupus erythematosus: prevalence, associations, and evolution // Am. J. Med. 2007. Vol. 120. P. 636.
-
Narchi H., Taylor R., Azmy A.F. et al. Shunt nephritis // J. Pediatr. Surg. 1988. Vol. 23. P. 839-841.
-
Neilson E.G. Pathogenesis and therapy of interstitial nephritis // Kidney Int. 1989. Vol. 35. P. 1257.
-
Neugarten J., Baldwin D.S. Glomerulonephritis in bacterial endocarditis // Am. J. Med. 1984. Vol. 77. P. 297-304.
-
Nolan C.M., Abernathy R.S. Nephropathy associated with methicillin therapy. Prevalence and determinants in patients with staphylococcal bacteremia // Arch. Intern. Med. 1977. Vol. 137. P. 997.
-
Ploier R., Geley, Syre G. The clinical picture in shunt nephritis // Wien. Klin. Wochenschr. 1985. Vol. 135. P. 311-315.
-
Ribera E., Miro J.M., Cortes E. et al. Influence of human immunodeficiency virus 1 infection and degree of immunosuppression in the clinical characteristics and outcome of infective endocarditis in intravenous drug users // Arch. Intern. Med. 1998. Vol. 158. P. 2043-2050.
-
Rose B.D. Pathophysiology of Renal Disease. 2nd ed. N.Y.: McGraw-Hill, 1987. 229 p.
-
Schoenbaum S.C., Gardner P., Shillitio J. Infections of cerebrospinal fluid shunts: epidemiology, clinical manifestations and therapy // J. Infect. Dis. 1975. Vol. 131. P. 543-552.
-
van der Meer J.T., Thompson J., Valkenburg H.A., Michel M.F. Epidemiology of bacterial endocarditis in The Netherlands. I. Patient characteristics // Arch. Intern. Med. 1992. Vol. 152. P. 1863-1868.
-
Vella J., Carmody M., Campbell E. et al. Glomerulonephritis after ventriculoatrial shunt // QJM. 1995. Vol. 88. P. 911-918.