Наследственные болезни : национальное руководство / Под ред. Н. П. Бочкова, Е. К. Гинтера, В. П. Пузырева - Москва : ГЭОТАР-Медиа, 2012. - 936 с. (Серия "Национальные руководства") - ISBN 978-5-9704-2231-1 |
Аннотация
Национальные руководства - первая в России серия практических руководств по основным медицинским специальностям, включающих всю основную информацию, необходимую врачу для непрерывного последипломного образования.
Национальное руководство "Наследственные болезни" содержит актуальную, современную информацию о геноме человека, общих вопросах медицинской генетики, клинической генетике. Руководство состоит из двух частей, в которых излагаются теоретические и клинические вопросы медицинской генетики. В первой части представлены новейшие данные по теоретическим вопросам медицинской генетики. Сведения об организации и функциях генома, генов и хромосом изложены в понятной для врачей форме, но без излишнего упрощения. Во второй части представлены вопросы клинической генетики, а именно методы диагностики наследственных болезней (от клинического уровня до секвенирования ДНК и РНК), принципов лечения и профилактики отдельных нозологических форм. Поскольку в национальных руководствах по другим специальностям описаны многочисленные наследственные болезни, на них можно найти ссылки [см. "Перечень наследственных болезней (синдромов), описание которых представлено в других национальных руководствах" на компакт-диске]. Приложение к руководству на компакт-диске включает более полную информацию по некоторым главам, электронную версию руководства, обширный иллюстративный материал, приложения, перечень наследственных болезней, описанных в других руководствах, фармакологический справочник. В подготовке настоящего издания в качестве авторов-составителей и рецензентов принимали участие ведущие ученые разных специальностей: генетики, иммунологи, невропатологи, фармакологи, онкологи и другие специалисты. Все рекомендации прошли этап независимого рецензирования.
Руководство предназначено для врачей-генетиков, врачей лаборантов-генетиков, врачей смежных специальностей, интернов, ординаторов, аспирантов, особенно по таким дисциплинам, как педиатрия, акушерство-гинекология, нервные болезни.
Гриф
Национальное руководство рекомендовано Российским обществом медицинских генетиков и Ассоциацией медицинских обществ по качеству
Глава 22. Хромосомные болезни
Введение
Хромосомные болезни занимают одно из ведущих мест в структуре врожденной и наследственной заболеваемости человека (табл. 22-1).
Таблица 22-1. Вклад конституциональных хромосомных аномалий в летальность и наследственные патологические изменения у человека (Gardner, Sutherland, 2004)
Эти расстройства обусловлены числовыми и структурными аномалиями хромосом, большинство из которых приводят к летальным эффектам. Лишь сравнительно немногие варианты нарушений кариотипа совместимы с постнатальным развитием организма и ведут к хромосомным болезням (Гинтер, 2003; Ворсанова и др., 2006; Бочков и др., 2010). До изложения характеристики этой группы болезней целесообразно рассмотреть основные положения, касающиеся спектра, механизмов формирования и закономерностей манифестации хромосомного дисбаланса. Все хромосомные аномалии разделяют на две группы - геномные и хромосомные мутации. К первым относят нарушения числа отдельных хромосом (анеуплоидия) и кратные изменения количества целых хромосомных наборов (полиплоидия). В категории хромосомных мутаций традиционно рассматривают структурные нарушения хромосом. По своему происхождению хромосомные аномалии могут быть наследуемыми и передаваться от родителей потомству (например, транслокации хромосом). Нарушения кариотипа могут возникать в ходе гаметогенеза de novo. Как правило, это числовые аномалии, формирующиеся в результате ошибок мейотической сегрегации хромосом. Ряд структурных аберраций также может возникать в гаметах (например, вследствие неаллельной гомологичной рекомбинации). Наследуемые и вновь возникшие гаметические мутации часто называют конституциональными хромосомными нарушениями, что связано с их присутствием во всех клетках организма. Хромосомные аномалии могут присутствовать и в соматических клетках. Если соматические мутации происходят на самых ранних этапах развития (например, на стадии дробления бластомеров), то с высокой вероятностью они будут представлены во всех клетках организма. Если же мутация возникнет на более поздних этапах онтогенеза (как правило, уже после обособления зародышевых листков), то она будет присутствовать только в части клеток организма. В этом случае хромосомная аномалия будет находиться в мозаичном состоянии. Следует отметить, что хромосомный мозаицизм может возникать не только при изначально нормальном кариотипе зиготы, но и быть результатом коррекции хромосомного нарушения гаметического происхождения. Подобные ситуации отмечают, например, при коррекции трисомии, в результате чего в развивающемся организме существуют популяции дисомных и трисомных клеток. Аномалии хромосом, связанные с нарушениями числа целых хромосомных наборов, у человека представлены триплоидией и тетраплоидией. Эти мутации летальные и, как правило, несовместимы с живорождением (Баранов, Кузнецова, 2007). Известны лишь единичные случаи рождения детей с множественными врожденными пороками развития при носительстве полиплоидного кариотипа. Продолжительность жизни таких детей обычно невелика, а сам факт прохождения пренатального периода онтогенеза зачастую можно объяснить присутствием в организме диплоидных клеток.
Типы геномных и хромосомных мутаций
Триплоидия - геномная мутация, при которой в кариотипе присутствуют три гаплоидных набора хромосом. У человека теоретически возможно существование трех вариантов триплоидных кариотипов: 69,XXX; 69,XXY и 69,XYY. Основной механизм формирования триплоидии - диспермное оплодотворение. Кроме того, она может возникнуть в результате слияния диплоидной и гаплоидной гамет, при этом формирование диплоидии в гамете может быть следствием нерасхождения целых хромосомных наборов в мейозе. Диплоидия в ооцитах возникает преимущественно в первом делении мейоза. В очень редких случаях триплоидный кариотип формируется в результате эндорепликации одного из родительских геномов в зиготе. Практически в 90% случаев дополнительный хромосомный набор в зиготе имеет отцовское происхождение, причем в 50-65% случаев он связан с диспермией. Теоретически ожидаемое соотношение частот кариотипов 69,XXX; 69,XXY и 69,XYY при условии формирования триплоидии вследствие диспермного оплодотворения должно составлять 1:2:1. В действительности оно стремится к соотношению 1:2. Это обусловлено тем, что кариотип 69,XYY обнаруживают очень редко, что связано с его ранним летальным эффектом.
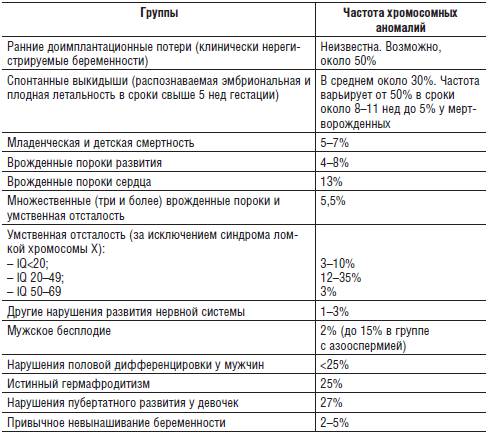
Фенотипические признаки триплоидии достаточно вариабельны. В одних случаях в материале спонтанных абортов обнаруживают пустые плодные мешки, в других - патологические плодные мешки с остатками резорбирующегося эмбриона, в третьих - плоды с множественными пороками развития. Известны редкие случаи рождения детей с триплоидным хромосомным набором (рис. 22-1). Такие новорожденные имеют небольшую массу тела, широкий задний родничок с недоразвитыми затылочными и теменными костями черепа, расщелину нёба, синдактилию III и IV пальцев рук, а также пороки сердца. В большинстве случаев их относят к триплоидно-диплоидным мозаикам. Вместе с тем описаны уникальные случаи рождения детей с мозаичными вариантами триплоидии, но без клона нормальных диплоидных клеток, например 69,XXX/47,XX,+8 (Yu et al., 1995) и 69,XXX/47,XX,+15 (Dean et al., 1997). Оба ребенка имели комплекс аномалий, характерных как для триплоидии, так и для трисомии по соответствующим хромосомам. Первая девочка прожила 58 дней, а вторая - 2,5 года.
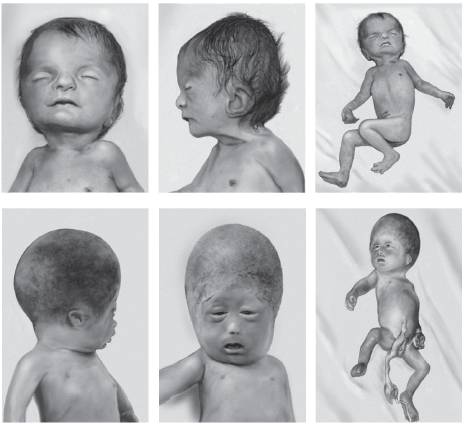
Рис. 22-1. Плоды с кариотипами 69,XXX (верхний ряд) и 69,XXY (нижний ряд). (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
При характеристике фенотипических признаков триплоидии в эмбриогенезе человека принципиально важно подчеркнуть, что ее эффекты во многом связаны с родительским происхождением дополнительного хромосомного набора вследствие эффекта геномного импринтинга. В случае триплоидного кариотипа диандрогенетического происхождения формируется клиническая картина частичного пузырного заноса, сопровождающаяся гиперплазией клеток трофобласта и кистозной трансформацией ворсин хориона. В случае отсутствия материнского хромосомного набора и при существовании двух гаплоидных наборов отцовского происхождения развивается полный пузырный занос, характеризующийся более выраженными гиперпролиферативными изменениями трофобласта, отсутствием сформированного эмбриона и высоким риском трансформации в хорионэпителиому. В связи с этим для прогноза риска развития онкологических осложнений у женщины существенное значение имеет своевременная цитогенетическая и молекулярно-генетическая диагностика пузырного заноса с целью дифференциации двух форм патологических изменений (Назаренко, 1993; рис. 22-2, см. цв. вклейку).
Тетраплоидия (присутствие в кариотипе четырех гаплоидных наборов хромосом) возникает преимущественно вследствие нарушений цитокинеза при дроблении бластомеров. Реже она является результатом слияния двух диплоидных гамет или оплодотворения яйцеклетки тремя гаплоидными сперматозоидами. Тетраплоидия - летальная мутация на организменном, но не на клеточном уровне. Так, например, известно, что ряд клеток (гепатоциты, кардиомиоциты, клетки эпителия мочевого пузыря и трофобласта плаценты) могут иметь не только тетраплоидный хромосомный набор, но и более высокие степени полиплоидизации. Фенотипически тетраплоидия часто ассоциирована с пустыми плодными мешками (анэмбриония). Ее присутствие на стадии бластомеров замедляет темпы их дробления и приводит к нарушению их миграции внутрь бластоцисты, что делает невозможным нормальное формирование и дифференцировку внутренней клеточной массы. Вместе с тем известны редкие случаи рождения детей с тетраплоидным хромосомным набором, в том числе и в чистой (немозаичной) форме. К настоящему времени в литературе описаны десять новорожденных с чистой формой тетраплоидии (Stefanova et al., 2010). Для большинства из них характерны внутриутробная задержка развития, гипотония, лицевые аномалии (выступающий лоб, микрофтальмия, низко посаженные уши, расщелина нёба), пороки сердца и нарушения психомоторного развития. Продолжительность жизни варьирует от 30 ч до 26 мес (рис. 22-3). Мозаичный вариант тетраплоидии имеет, как правило, более мягкое фенотипическое проявление, что может быть связано с присутствием полиплоидии только в части клеток или тканей (рис. 22.4).
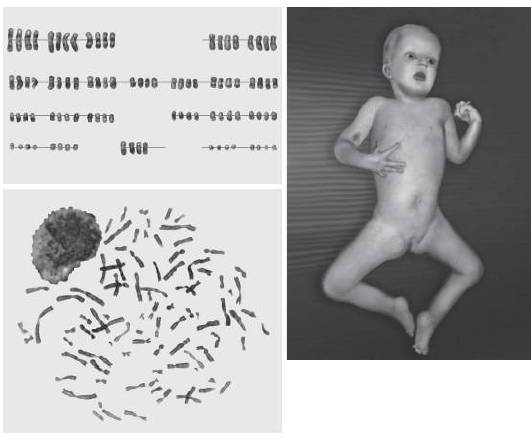
Рис. 22-3. Чистый вариант тетраплоидии у пациента в возрасте 26 мес [Guc-Scekic M., Milasin J., Stevanovic M. et al. Tetraploidy in a 26-month-old girl (cytogenetic and molecular studies) // Clin.Genet. - 2002. - Vol. 61. - P. 62-65]
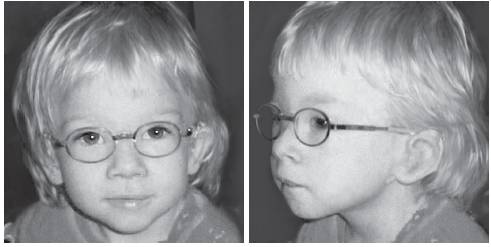
Рис. 22-4. Пациент с мозаичным вариантом тетраплоидии в лимфоцитах периферической крови в возрасте 2 лет и 7 мес. Кариотип: 92,XXXX[45]/46,XX[55]. Кариотип клеток буккального эпителия ротовой полости и фибробластов кожи нормальный (Stefanova I., Jenderny J., Kaminsky E. et al. Mosaic and complete tetraploidy in live-born infants: two new patients and review of the literature // Clin. Dysmorphol. - 2010. - Vol. 19. - P. 123-127)
Анеуплоидия - наиболее распространенная геномная мутация у человека. Среди всех клинически распознаваемых беременностей частота ее обнаружения составляет около 5% (Hassold, Hunt, 2001). Теоретически в кариотипе человека анеуплоидия может быть представлена следующими формами:
-
нуллисомия - полное отсутствие гомологов какой-либо пары хромосом;
-
трисомия - присутствие в кариотипе дополнительной гомологичной хромосомы;
-
тетрасомия и полисомия - присутствие четырех и более копий гомологичных хромосом;
-
двойные и тройные трисомии - наличие в кариотипе дополнительных копий двух или трех пар гомологичных хромосом одновременно.
Большинство этих вариантов приводит к нарушениям внутриутробного развития. Нередко их обнаруживают при исследовании материала спонтанных абортов или в бластомерах при проведении преимплантационной генетической диагностики. Только немногие варианты анеуплоидий (трисомии по некоторым аутосомам и половым хромосомам, моносомия по хромосоме Х) совместимы с живорождением и приводят к формированию клинически хорошо очерченных хромосомных болезней. Самая частая геномная мутация у человека среди всех типов анеуплоидий - трисомия по аутосомам. Среди спонтанных абортусов I триместра беременности на ее долю приходится около 50% регистрируемых хромосомных нарушений. Частота трисомий по разным хромосомам существенно варьирует (Griffin, 1996). Так, самая распространенная анеуплоидия - трисомия хромосомы 16, обнаруживаемая у 30% эмбрионов с трисомным хромосомным набором. Далее следуют трисомии по акроцентрическим хромосомам и хромосомам 2, 7 и 18. Трисомии по хромосомам 1, 5 и 19, напротив, чрезвычайно редки. Их практически не обнаруживают в материале спонтанных абортов, так как они приводят к остановке развития на самых ранних этапах эмбриогенеза. Очевидно, что наблюдаемые различия в частоте трисомий отражают разную морфофункциональную значимость генного состава хромосом, воввлеченных в анеуплоидию. В основе возникновения трисомии лежат преимущественно ошибки сегрегации хромосом в мейотическом делении клеток, приводящие к формированию дисомных гамет. Среди них следует отметить нерасхождение гомологичных хромосом в первом делении или сестринских хроматид во втором делении мейоза, преждевременную сегрегацию хроматид в первом мейозе и нарушения кроссинговера, приводящие к ахиазматическому нерасхождению гомологов. Большинство ошибок сегрегации хромосом возникает в первом делении мейоза у женщины. Более того, вероятность формирования анеуплоидных гамет заметно увеличивается с возрастом матери, что считают одним из основных показаний для проведения пренатальной диагностики хромосомных болезней.
Двойные и тройные трисомии - редкий тип аномалий, формирующийся вследствие одновременного нерасхождения двух или трех хромосом. Частота их обнаружения в материале спонтанных абортов составляет 0,2-2,8 и 0,025% соответственно (Reddy, 1997, 1999). Есть данные о более раннем прекращении развития зародышей с двойными трисомиями и о более старшем возрасте их матерей по сравнению с таковым при трисомиях по одной хромосоме. У живорожденных описаны двойные трисомии хромосом 13, 18, 21, X и Y. Известны тетрасомии по половым хромосомам, а также по хромосоме 21.
Моносомия - геномная мутация с очень ранним летальным эффектом. Элиминация большинства эмбрионов с аутосомными моносомиями происходит уже на доимплантационном этапе развития. Среди спонтанных абортусов I триместра беременности частота моносомий по аутосомам составляет менее 1% всех регистрируемых нарушений кариотипа. Как правило, это моносомии акроцентрических хромосом, преимущественно 21 и 22. Следует отметить, что применение методов молекулярно-цитогенетического анализа (FISH, CGH) для исследования кариотипа клеток спонтанных абортусов (особенно при невозможности получения препаратов метафазных хромосом) позволило обнаружить более широкий спектр аутосомных моносомий (Daniely et al., 1999; Lebedev et al., 2004). Присутствуя, как правило, в мозаичном состоянии с нормальной клеточной линией, такие хромосомные нарушения оказываются совместимыми с прохождением ранних постимплантационных этапов развития. Более того, известны уникальные случаи рождения детей с моносомией 21 (Garzicic et al., 1988).
Единственной совместимой с жизнью считают моносомию по хромосоме Х, которую обнаруживают среди новорожденных с частотой 0,04-0,07%. В 70-80% случаев единственная хромосома Х наследуется от матери, а отцовские хромосомы Х или Y теряются в гаметогенезе или на ранних этапах дробления. Существование в кариотипе моносомии Х приводит к формированию синдрома Шерешевского-Тернера. Тем не менее летальность при этом хромосомном нарушении все же велика и приближается к 100%. Большинство эмбрионов с моносомией Х погибают до 6-й недели гестации. В 60% случаев зародыши представлены пустыми плодными мешками. Остальные эмбрионы в эти сроки не имеют видимых анатомических аномалий. На более поздних этапах формируется комплекс патоморфологических нарушений: генерализованный отек, билатеральная гигрома шеи, пороки сердца, аорты, мочеполовой системы, аномалии скелета, узловатый амнион и аплазия сосудов пуповины. Моносомия по хромосоме Х сопровождается нарушением формирования гонад, которое выражается в значительной редукции фолликулов и их замене соединительной тканью.
Нуллисомии по аутосомам регистрируют только у эмбрионов на преимплантационных этапах развития.
Хромосомные мутации - нарушения структуры хромосом. В основе их возникновения лежат разрывы сахарофосфатных связей в молекуле ДНК с вовлечением одной или нескольких хромосом. Воссоединение разорванных концов происходит с помощью ферментов репарации ДНК. Если концы фрагментов, принадлежащих к одной и той же хромосоме, объединятся без потери хромосомного материала и в прежней ориентации, то хромосома сохранит свою структуру. В случае утраты хромосомного материала, изменения ориентации фрагмента или его присоединения к другим негомологичным хромосомам будут возникать структурные перестройки. Существует несколько типов структурных аберраций хромосом.
Делеция - утрата части хромосомного материала. Она может возникать как на концевых участках хромосом (терминальная делеция), так и во внутрихромосомных регионах (интерстициальная делеция). Фенотипический эффект делеций обусловлен потерей хромосомного материала, содержащего определенный набор генов. Дополнительный вклад может вносить нарушение структурной последовательности гена, непосредственно вовлеченного в разрыв хромосомы. В некоторых случаях размер делеций может составлять всего несколько миллионов пар азотистых оснований (1-3 Mb), что значительно затрудняет их цитогенетическую диагностику с использованием дифференциально окрашенных препаратов метафазных хромосом и требует применения высокоразрешающих методов молекулярно-цитогенетического анализа. Кроме того, использование современных молекулярно-цитогенетических технологий, и особенно микрочипов, не только позволяет довольно успешно диагностировать уже известные микроделеционные синдромы, но и служит основой для описания новых хромосомных болезней (Slavotinek, 2008).
В процессе мейоза при конъюгации гомологичных хромосом, одна из которых несет интерстициальную делецию, интактный участок неповрежденной хромосомы не может найти в первой гомологичную последовательность для взаимодействия. В результате образуется петлевидная структура, которая помешает нормальному течению рекомбинации и в конечном итоге может привести к нарушению сегрегации хромосом (рис. 22-5, см. цв. вклейку). Таким образом, у носителей делеций высоковероятны нарушения мейоза, сопровождающиеся формированием несбалансированных или анеуплоидных гамет.
Дупликация - хромосомная мутация, представленная удвоением участка хромосомы. Если он располагается непосредственно за исходной последовательностью, то такую дупликацию называют тандемной. Фенотипический эффект дупликаций связан с увеличением копийности генов, расположенных в регионе мутации. Так же как и при делеции, хромосома с дупликацией неспособна к нормальному взаимодействию с другим гомологом в ходе мейоза, что приводит к образованию петлевых структур и, как следствие, нарушениям сегрегации и формированию несбалансированных гамет (рис. 22.6, см. цв. вклейку). При характеристике этого типа мутаций особого внимания заслуживают так называемые сегментные дупликации генома - повторяющиеся блоки хромосом с высокой (более 95%) степенью идентичности нуклеотидных последовательностей. Неаллельная гомологичная рекомбинация между ними приводит к формированию микроделеций и микродупликаций. В зависимости от размера вовлеченного региона эти перестройки приводят к развитию либо микроделеционных (микродупликационных) синдромов, либо моногенных заболеваний.
Инверсия - поворот участка хромосомы на 180?. В зависимости от того, затрагивает ли она одно или оба плеча хромосомы, различают пара- и перицентрические инверсии соответственно. Течение мейоза хромосом с инверсией значительно затруднено вследствие образования особых петлевых структур (рис. 22-7, 22-8, см. цв. вклейку). При сегрегации гомологичных хромосом, одна из которых несет инверсию, вероятность формирования нормальных гамет составляет только 25%. В связи с этим носители инверсии, у которых она может не иметь никаких фенотипических признаков, характеризуются нарушениями гаметогенеза вследствие высокой вероятности формирования несбалансированных гамет.
Инсерция - вставка фрагмента одной хромосомы в другой регион той же или другой негомологичной хромосомы. В первом случае образуется интрахромосомная, во втором - интерхромосомная инсерция. В случае если встраиваемый фрагмент хромосомы имеет терминальное или интерстициальное происхождение, для возникновения инсерции требуется три или четыре хромосомных разрыва соответственно. В зависимости от ориентации встроенного фрагмента по отношению к центромере различают прямые и инвертированные инсерции. При инсерции не происходит изменения числа копий перестроенных участков хромосом, а фенотипический эффект может быть связан с эффектом положения, а именно с перемещением блока структурных генов из одного хромосомного окружения в другое. Поведение хромосом с инсерцией в мейозе в значительной степени зависит от размера встроенного фрагмента и характеризуется конъюгацией гомологичных хромосом при инсерциях небольшого размера либо формированием квадривалентов при значительных вставках. Результатом мейоза может стать образование гамет с сегментными анеуплоидиями. Именно поэтому носительство инсерций сопровождается серьезными репродуктивными проблемами. Вероятность рождения ребенка с несбалансированным хромосомным набором для мужчин-носителей инсерции составляет около 32%, а для женщин - 36% (Gardner, Sutherland, 2004). Риск увеличивается при носительстве инсерций небольшого размера, и наоборот, снижается для носителей крупных инсерций. Среди фенотипически нормального потомства около половины индивидуумов будут иметь нормальные хромосомы, тогда как вторую половину составят гетерозиготные носители инсерции.
Транслокация - перенос участка одной хромосомы на другую хромосому. Чаще всего в транслокацию вовлечены две хромосомы, но возможны перестройки с участием трех или более хромосом. В отдельный класс выделяют так называемые робертсоновские транслокации, образующиеся вследствие разрыва и воссоединения двух акроцентрических хромосом в области центромеры. Если при обмене не происходит потери хромосомного материала, такие транслокации называют сбалансированными. Взаимный обмен участками двух разных гомологичных хромосом приводит к формированию реципрокной транслокации. Носительство в кариотипе реципрокных и робертсоновских транслокаций часто не сопровождается какимилибо клиническими признаками. При несбалансированных транслокациях возникают фенотипические нарушения, связанные с дисбалансом числа копий участка хромосомы, вовлеченного в перестройку. В таком случае в кариотипе возникают сегментные анеуплоидии, представленные частичной моносомией или частичной трисомией по хромосомному региону, вовлеченному в транслокацию.
Вследствие того что при транслокациях формируется хромосома, представленная участками двух разных хромосом, прохождение такой перестроенной хромосомой мейотического деления имеет свои особенности. Так, в конъюгации принимает участие уже не одна, а две пары гомологичных хромосом. В ходе обмена формируется особая фигура, получившая название «транслокационный крест». Только один из шести возможных вариантов сегрегации хромосом, составляющих транслокационный крест, ведет к формированию сбалансированных гамет (рис. 22-9, см. цв. вклейку). В связи с этим у носителей хромосомных транслокаций высока вероятность нарушений мейоза и формирования несбалансированных и анеуплоидных гамет (рис. 22-10, см. цв. вклейку), что приводит к бесплодию или невынашиванию беременности.
Изохромосома - хромосома, формирующаяся вследствие аномального поперечного деления центромеры, что ведет к разделению короткого и длинного плеча. После дупликации одного из плеч возникает изохромосома по короткому или длинному плечу. Фенотипический эффект у носителей таких перестроек связан как с отсутствием нуклеотидных последовательностей одного из плеч, так и с дупликацией хромосомного материала другого плеча.
Дицентрическая хромосома возникает в результате воссоединения двух поврежденных хромосом, сохранивших свои центромерные последовательности.
Иногда образование дицентрической хромосомы является следствием неравного кроссинговера между блоками сегментных дупликаций. В тех случаях, когда последние находятся в инвертированном положении относительно друг друга и располагаются вблизи центромерных регионов хромосом, неаллельная гомологичная рекомбинация между ними будет приводить к формированию изодицентрических хромосом - изохромосом с двумя центромерными регионами (рис. 22-11, см. цв. вклейку). Именно по такому механизму образуется одна из наиболее распространенных маркерных хромосом, обнаруживаемых у новорожденных с множественными врожденными пороками развития, - изодицентрическая хромосома 15, имеющая в своем составе две центромерные последовательности.
Кольцевая хромосома возникает при утрате обоих теломерных участков одной хромосомы с воссоединением ее концов. Это нестабильная структура, часто подверженная потере при клеточном делении. Фенотипический эффект такой перестройки может быть связан с утратой части хромосомного материала при формировании кольцевой хромосомы, а также с самой ее потерей в части соматических клеток.
Классификация хромосомных болезней
В основу классификации патологических изменений хромосом может быть положено несколько принципов (Бочков и др., 2010). Первый принцип (этиологический) связан с характеристикой типа мутации. В общем виде клиническая картина хромосомного заболевания складывается из двух составляющих: хромосомной структуры (сегмент, плечо или целая хромосома), вовлеченной в аберрацию, и характера генетического нарушения (уменьшение или увеличение числа копий хромосомного материала).
Второй принцип связан с определением типа клеток, в которых возникла мутация, - в гаметах или соматических клетках на постзиготических этапах развития. Гаметические мутации формируются вследствие нарушений мейоза. Это могут быть геномные мутации, например нерасхождение хромосом в первом или во втором мейотическом делении, приводящее к возникновению анеуплоидных гамет и впоследствии анеуплоидных (трисомных или моносомных) зигот. Структурные хромосомные аберрации также могут формироваться в мейозе вследствие нарушений конъюгации хромосом и генетической рекомбинации. В случае гаметической мутации конституциональное хромосомное нарушение будет присутствовать во всех клетках организма.
Существование хромосомных нарушений в зиготе может кардинальным образом нарушить всю программу развития. Показательна в этом отношении трисомия по хромосоме 8. Эта анеуплоидия представляет наглядный пример влияния происхождения хромосомной аномалии на развитие организма. Трисомия по хромосоме 8 мейотического происхождения несовместима с нормальным эмбриогенезом и приводит к спонтанному прерыванию беременности. Если же эта мутация возникнет вследствие митотического нерасхождения хромосом, то она может быть совместима с пренатальным развитием и живорождением, сопровождаясь формированием ряда пороков. Следует отметить, что большинство дифференцированных тканей человека толерантны к достаточно большому числу клеток с трисомией хромосомы 8. Так, у новорожденных с синдромом трисомии по хромосоме 8 эту аномалию можно обнаружить в 100% лимфоцитов. Предложен ряд гипотез, объясняющих такие эффекты трисомии по хромосоме 8 (Robinson et al., 1999). Одна из них предполагает, что подобная анеуплоидия мейотического происхождения - одна из наиболее стабильных хромосомных аномалий, и ее коррекция происходит сравнительно редко. Другая гипотеза допускает, что негативное влияние трисомии по хромосоме 8 на развитие может быть более выраженным, если она затрагивает рано дифференцирующиеся ткани, к которым относят и трофобласт. Это возможно только при мейотическом происхождении, которое приводит к присутствию трисомии уже в зиготе. Предполагают, что существование этого нарушения может ингибировать ключевые этапы дифференцировки трофобласта. Вероятно, если оно возникнет на более поздних стадиях онтогенеза, то его эффекты не будут столь значительными.
Таким образом, соматические мутации могут возникать в зиготе в период внутриутробного развития или в постнатальном онтогенезе. В результате в организме присутствуют два или более клеточных клонов с разными хромосомными наборами, при этом, как правило, один клон клеток имеет нормальный кариотип, а другой - аномальный. Такие варианты хромосомных аномалий называют мозаичными. В редких случаях комбинация клеток с разными типами хромосомных нарушений может быть следствием не мозаицизма, а химеризма - объединения бластомеров, принадлежащих разным зародышам, на самых ранних этапах развития. Клинические признаки мозаичных хромосомных аномалий зависят от сочетания множества факторов, в том числе от типа мутации, вовлеченной хромосомы или ее сегмента, числа клеток с аномальным кариотипом и их распределения в различных тканях организма. Как правило, строгой корреляции между этими показателями и степенью выраженности клинических признаков при хромосомном мозаицизме нет.
Наконец, третий принцип классификации хромосомных болезней связан с установлением поколения, в котором возникла мутация: появилась ли она в гаметах здоровых родителей de novo (спорадические случаи) или родители уже были носителями указанной хромосомной аномалии. О наследуемых хромосомных болезнях говорят в том случае, когда хромосомная мутация уже присутствует в клетках одного из родителей и может быть передана потомству через гаметы. Обычно такие ситуации характерны для носителей структурных перестроек хромосом (например, сбалансированных или робертсоновских транслокаций). Наследование хромосомного нарушения также может быть результатом гонадного мозаицизма - явления, при котором у здорового индивидуума с нормальным хромосомным набором в гонадах есть клон клеток с хромосомной аберрацией. Как правило, это числовые хромосомные нарушения, сохранившиеся в зародышевой половой линии после коррекции анеуплоидии в соматических клетках развивающегося эмбриона. Другим фактором, приводящим к формированию гонадного мозаицизма, могут быть ошибки сегрегации хромосом, возникающие на стадии митотического деления клеток-предшественниц гамет.
Таким образом, для диагностики хромосомного заболевания и прогноза его повторного возникновения в семье необходимо определить тип мутации, вовлеченную хромосому, форму мутации (полная или мозаичная) и ее встречаемость в родословной (спорадический или наследуемый случай). Учитывая эти особенности, следует также отметить, что для установления точного диагноза не всегда бывает достаточно обследования самого пациента. Иногда требуется проведение цитогенетического исследования его родителей и сибсов.
Патогенез хромосомных болезней
В основе патогенеза хромосомных заболеваний лежит дисбаланс по числу копий хромосомного материала. При анеуплоидиях в него вовлечено большое число генов, что в значительной степени затрудняет анализ ключевых механизмов развития заболевания. При некоторых структурных хромосомных мутациях дополнительный вклад в формирование клинической картины может вносить эффект положения. Несмотря на достаточно хорошую клиническую изученность хромосомных синдромов и идентификацию аномалий хромосом, определяющих их, конкретные механизмы развития заболеваний остаются понятными лишь в общих чертах. Так, при оценке эффектов хромосомного дисбаланса при полных или сегментных анеуплоидиях предложено выделять три типа генетических эффектов: специфические, полуспецифические и неспецифические (Бочков и др., 2010).
Специфические эффекты связаны с изменением числа структурных генов: при трисомиях оно увеличивается, а при моносомиях, наоборот, уменьшается. Вполне ожидаемо, что вслед за изменением числа копий хромосомного материала должно меняться и число кодируемых белковых продуктов. Тем не менее в действительности при числовых хромосомных нарушениях часто не происходит строго пропорционального изменения степени экспрессии генов, что объясняют разбалансировкой сложных регуляторных процессов в клетке. Более того, при хромосомных синдромах происходит существенное изменение числа и активности других белков, гены которых локализованы на других хромосомах, не вовлеченных в нарушение.
При исследованиях, проведенных среди больных с синдромом Дауна, в зависимости от изменения активности при трисомии были идентифицированы три группы генов, расположенных на хромосоме 21 (Prandini et al., 2007). В первую группу вошли гены, степень экспрессии которых значительно превышает уровень активности в дисомных клетках. Предполагают, что именно они обусловливают формирование основных клинических признаков синдрома Дауна у большинства пациентов. Вторую группу составили гены, степень экспрессии которых частично перекрывается с уровнем их активности при нормальном кариотипе. Как полагают, они определяют формирование вариабельных признаков синдрома, отмечаемых не у всех пациентов. Наконец, в третью группу вошли гены, уровень экспрессии которых в трисомных и дисомных клетках практически не различался. Вероятно, они в малой степени вовлечены в формирование клинических признаков заболевания. Следует отметить, что к первым двум группам были отнесены только 60% генов, локализованных на хромосоме 21 и экспрессирующихся в лимфоцитах, и 69% генов, экспрессирующихся в фибробластах (табл. 22-2). На рис. 22-12 представлена «фенотипическая карта» хромосомы 21 с указанием сцепления некоторых клинических признаков синдрома Дауна с определенными хромосомными регионами. Данная карта построена на основе анализа фенотипического проявления сегментных трисомий по тем или иным регионам хромосомы 21.
Таблица 22-2. Некоторые дозозависимые гены, определяющие формирование основных клинических признаков синдрома Дауна при трисомии 21
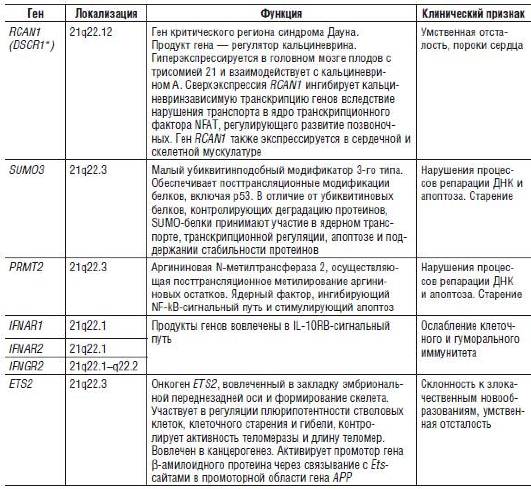
* DSCR (Down syndrome critical region) - критический регион синдрома Дауна.
Полуспецифические эффекты могут быть обусловлены изменением числа высококопийных генов. К ним относят гены рибосомных и транспортных РНК, гистоновых и рибосомных белков.
Неспецифические эффекты связаны с изменением числа гетерохроматиновых последовательностей, структурно-регуляторная функция которых хорошо известна.
С клинической точки зрения общим для всех форм хромосомных болезней считают поражение множества систем органов. При каждой форме заболевания могут возникать несколько различных фенотипических отклонений, часто перекрывающихся при разных синдромах. Лишь ограниченное число синдромов характеризуется строго определенной комбинацией признаков. Множественность поражений при развитии хромосомного заболевания объясняют существованием хромосомной аномалии во всех клетках организма, начиная с самых первых этапов развития. Присутствие мутации значительно нарушает всю программу онтогенеза, приводя к нарушению морфогенетических процессов в ключевые периоды внутриутробного развития.
В основе формирования клинической картины хромосомного заболевания лежит сочетание нескольких основных факторов:
-
индивидуальность вовлеченной в аномалию хромосомы или ее участка (специфический набор генов);
-
тип мутации и определяемый ею дозовый эффект (увеличение или уменьшение числа копий, полная или сегментная анеуплоидия);
-
размер недостающего (при частичных моносомиях) и избыточного (при частичных трисомиях) участков хромосомы;
-
непосредственные структурные повреждения генов при разрывах хромосом;
-
позиционный эффект, связанный с переносом генов в новое окружение;
-
полная или мозаичная форма хромосомного нарушения, степень распространения аномального клеточного клона в пределах ткани и его межтканевое распределение;
-
родительское происхождение хромосомных аномалий, связанное с эффектом геномного импринтинга;
-
генотип организма, оказывающий модифицирующее влияние на клиническую картину хромосомного нарушения;
-
условия окружающей среды, в которой протекает развитие организма с хромосомным нарушением.
Эти сочетания определяют клинический полиморфизм хромосомных заболеваний. Вариабельность фенотипических признаков геномных и хромосомных мутаций может быть достаточно широкой: от ранней эмбриональной гибели до рождения ребенка с хромосомной болезнью. Спектр эффектов конституциональных хромосомных аномалий можно классифицировать следующим образом (Schinzel, 2001):
-
нарушение бластогенеза с аномальной имплантацией или невозможностью имплантации бластоцисты;
-
нарушение эмбриогенеза со спонтанным прерыванием беременности (обычно в I триместре);
-
серьезные нарушения внутриутробного морфогенеза, приводящие к мертворождениям или ранней неонатальной гибели;
-
нарушения внутриутробного морфогенеза различной степени тяжести с рождением жизнеспособного организма;
-
минимальные фенотипические признаки хромосомного дисбаланса или их отсутствие.
Как было отмечено ранее, степень выраженности клинического признака при хромосомном заболевании определяется сочетанием нескольких факторов и в зависимости от вклада цитогенетических нарушений коррелирует с размером вовлеченного в перестройку хромосомного региона, увеличением или уменьшением числа его копий и уровнем внутри- и межтканевого мозаицизма. При дупликациях аутосомных сегментов степень выраженности нарушений роста, умственной отсталости и частота пороков развития обычно прямо связаны с размером дуплицированного сегмента, за исключением аутосомных трисомий, большинство которых несовместимо с живорождением. Дупликации определенного хромосомного сегмента, как правило, ведут к менее тяжелым фенотипическим последствиям, чем делеции.
Часто формирование какого-либо патологического признака связано с изменением копийности определенных районов хромосом, возникающим вследствие делеций и дупликаций. В таких случаях детальный анализ клинической картины при хромосомных заболеваниях позволяет провести картирование генов, ответственных за возникновение того или иного нарушения. Как правило, клинические признаки, связанные с дупликацией хромосомного участка, можно отметить и при заболеваниях, возникающих при полных трисомиях соответствующих хромосом.
Хромосомные болезни, обусловленные нарушениями числа хромосом
С клинической точки зрения числовые нарушения аутосом характеризуются следующими основными признаками:
Хотя присутствие любой из этих четырех групп признаков не считают обязательным при том или ином синдроме, умственная отсталость - одно из наиболее типичных нарушений.
Синдром Дауна (трисомия хромосомы 21). Наиболее распространенное хромосомное заболевание. Популяционная частота составляет 1:600-700 новорожденных. Это первый синдром, хромосомная этиология которого была установлена Ж. Леженом и соавт. в 1959 г. Цитогенетические варианты синдрома Дауна разнообразны. Основную долю (до 95%) составляют случаи полной трисомии 21, возникающие вследствие нерасхождения хромосом в мейозе. Вклад материнского нерасхождения в гаметические формы болезни составляет 85-90%, а отцовского - только 10-15%. Примерно 75% нарушений возникают в первом делении мейоза у матери и только 25% - во втором. Около 2% детей с синдромом Дауна имеют мозаичные формы трисомии 21 (47,+21/46). Примерно 3-4% больных имеют транслокационную форму трисомии по типу робертсоновских транслокаций между акроцентрическими хромосомами (D/21 и G/21). Около одной четверти транслокационных форм наследуются от родителей-носителей, тогда как три четверти их возникают de novo.
Основные клинические признаки синдрома (см. рис. 22-12): типичное плоское лицо, брахицефалия, аномалии глаз (монголоидный разрез глаз, эпикант, пятна Брушфильда, ранняя катаракта, миопия), открытый рот, аномалии зубов, короткий нос, плоская переносица, избыток кожи на шее, короткие конечности, поперечная четырехпальцевая ладонная складка, широкий промежуток между I и II пальцами стопы. Из пороков внутренних органов часто отмечают врожденные пороки сердца (дефекты межжелудочковой и межпредсердной перегородки, открытый артериальный проток) и ЖКТ, которые в значительной степени определяют продолжительность жизни пациентов с синдромом Дауна. Большинство больных страдают умеренной или тяжелой степенью умственной отсталости. Более мягкие фенотипические признаки характерны для пациентов с мозаичными формами синдрома (рис. 22-13).
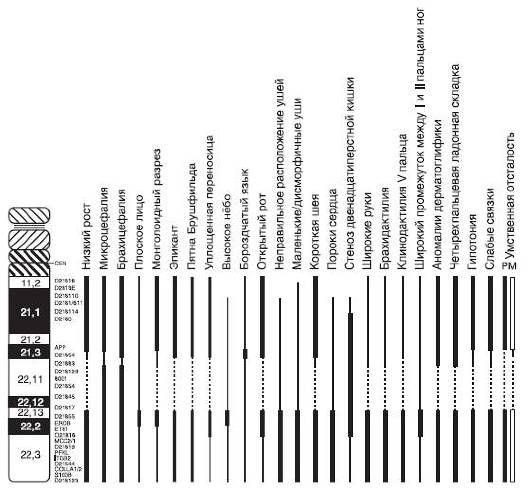
Рис. 22-12. «Фенотипическая карта» хромосомы 21 (Korenberg J.R., Chen X.-N., Schipper R. et al. Down syndrome phenotypes: The consequences of chromosome imbalance // PNAS. - 1994. - Vol. 91. - P. 4997-5001). Р, М - отцовский и материнский гомологи
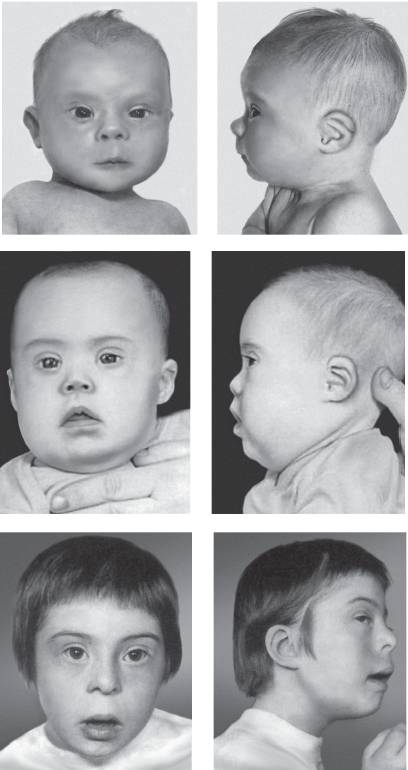
Рис. 22-13. Фенотип пациентов с синдромом Дауна в возрасте 5 нед (верхний ряд), 3,5 года (средний ряд) и 7 лет (нижний ряд) (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
Синдром Патау (трисомия хромосомы 13). Хромосомная этиология заболевания впервые была описана К. Патау в 1960 г. Популяционная частота варьирует в диапазоне 1:7800-14 000. Заболевание возникает преимущественно вследствие трисомии хромосомы 13, как правило, материнского происхождения. Кроме того, развитие синдрома может быть связано с транслокационными вариантами (робертсоновские транслокации), мозаичными формами, дополнительной кольцевой хромосомой 13 и изохромосомами.
Клинически синдром Патау (рис. 22-14) характеризуется микроцефалией, расщелинами верхней губы и нёба, низко посаженными деформированными ушными раковинами, микрогенией, гипотелоризмом, дисплазией сетчатки, полидактилией, поперечной ладонной складкой и множественными пороками внутренних органов: врожденными пороками сердца (дефекты перегородок и крупных сосудов), незавершенным поворотом кишечника, поликистозом почек и удвоением мочеточника. Обнаруживают крипторхизм, гипоплазию наружных половых органов, удвоение матки и влагалища. Для детей характерна глубокая идиотия. Продолжительность жизни, как правило, составляет 2-3 мес и редко достигает одного года.
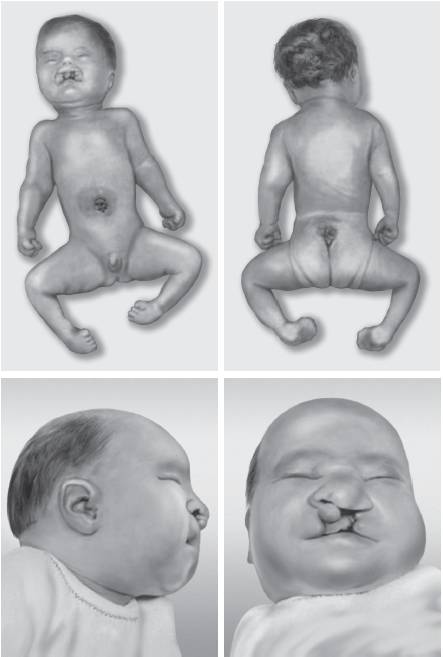
Рис. 22-14. Новорожденные с синдромом Патау (трисомия хромосомы 13) (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
Синдром Эдвардса (трисомия хромосомы 18). Впервые описан Эдвардсом в 1960 г. Популяционная частота составляет 1:6000-8000 случаев. Второе по распространенности после синдрома Дауна хромосомное заболевание. Большинство случаев (90%) связано с полной формой хромосомы 18, возникающей вследствие ошибок первого деления мейоза у матери. Транслокационные варианты регистрируют крайне редко. Критический регион, ответственный за формирование основных клинических признаков синдрома, - сегмент 18q11.
Новорожденные с синдромом Эдвардса имеют малую массу тела. Основные диагностические признаки заболевания: долихоцефалия, гипертелоризм, низко посаженные уши аномальной формы, микрогнатия, микростомия и скошенный подбородок (рис. 22-15). Возможны аномалии развития конечностей, отсутствие дистальной складки на мизинце и гипоплазия ногтей. Из пороков внутренних органов характерными считают комбинированные пороки сердечно-сосудистой системы, незавершенный поворот кишечника, пороки развития почек и крипторхизм. Отмечают задержку психомоторного развития, идиотию, имбецильность. Продолжительность жизни обычно не превышает одного года.
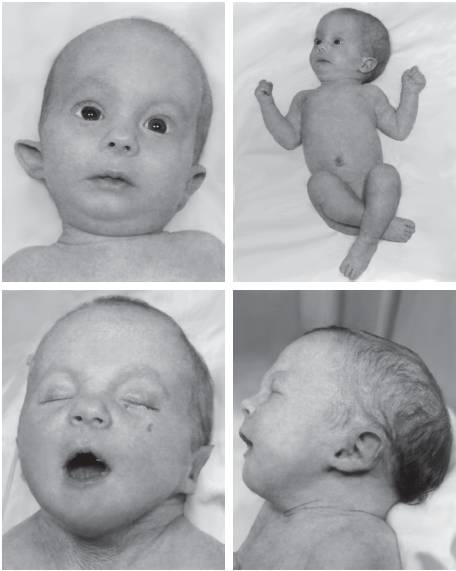
Рис. 22-15. Новорожденные с синдромом Эдвардса (трисомия хромосомы 18). (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.). Окончание - на с. 572
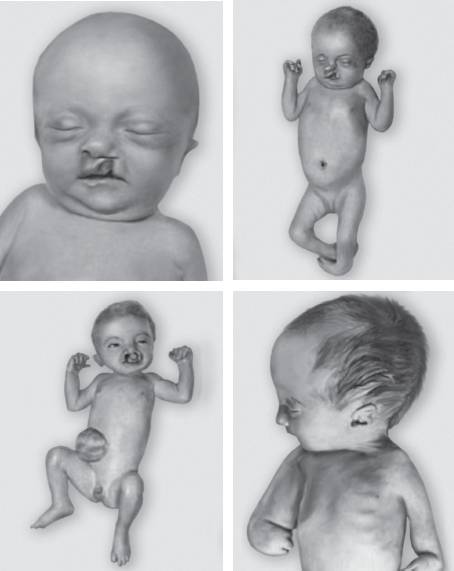
Рис. 22-15. ОкончаниеТрисомии по хромосомам 8, 9 и 14 среди новорожденных регистрируют редко. Описаны единичные случаи некоторых трисомий.
Синдром трисомии по хромосоме 8. Впервые описан в 1962 г. Редкое заболевание, частота которого в популяции составляет 1:50 000. Возникает в результате хромосомного нерасхождения в соматических клетках на ранних стадиях развития. Трисомия 8 гаметического происхождения характеризуется, как было отмечено выше, ранней эмбриолетальностью. У новорожденных обнаруживают как полные, так и мозаичные формы трисомии, при этом корреляция между распространенностью анеуплоидного клона и тяжестью заболевания обычно отсутствует.
Основные диагностические признаки синдрома: макроцефалия, микрогнатия, массивный выступающий лоб, широкая спинка носа и большие оттопыренные уши (рис. 22-16). Среди аномалий скелета обнаруживают добавочные ребра и позвонки, закрытые спинномозговые грыжи в шейном и грудном отделе позво ночника, аплазию и гипоплазию надколенника, а также короткую шею. Отмечают множественные контрактуры суставов, клинодактилию и камптодактилию. Среди пороков внутренних органов распространены аномалии мочеполовой (гидронефроз) и сердечно-сосудистой системы (дефекты перегородок и крупных сосудов). У больных отмечают задержку психомоторного и речевого развития. Интеллект обычно снижен.
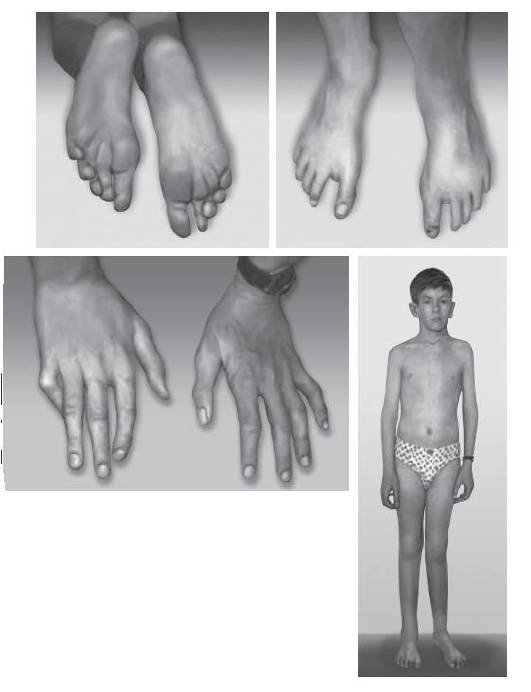
Рис. 22-16. Фенотип пациента в возрасте 15 лет с мозаичным вариантом трисомии 8 (Wiśniewska M., Mazurek M. Trisomy 8 mosaicism syndrome // J. Appl. Genet. - 2002. - Vol. 43. - P. 115-118) Синдром трисомии по хромосоме 9. Впервые описан в 1970 г. В большинстве случаев заболевание обусловлено нерасхождением хромосом на ранних стадиях развития, и только иногда оно связано с гаметическими мутациями. Возможны полные и мозаичные формы (рис. 22-17). Основные клинические признаки: долихоцефалия, глубоко посаженные глаза, высокий лоб, широкая переносица, высокое нёбо (часто с расщелиной), микроретрогнатия, деформация ушных раковин и короткая шея. Отмечают аномалии скелета, включающие дисплазию тазобедренного сустава, вывих локтевого или коленного сустава, патологические изменения ребер. Из пороков внутренних органов типичны аномалии сердечно-сосудистой, мочеполовой систем и ЖКТ. Большинство носителей трисомии 9 погибают в первые 4 мес жизни, преимущественно от респираторных инфекций.
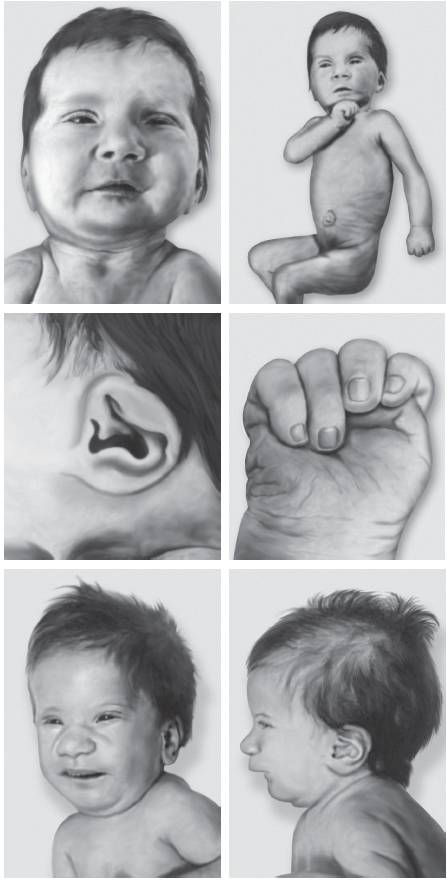
Рис. 22-17. Фенотипическое проявление мозаичных форм трисомии 9 (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
Синдром трисомии по хромосоме 14. Впервые описан в 1975 г. В основном представлен мозаичными формами (рис. 22-18) и робертсоновскими транслокациями 14/14. Основные диагностические признаки: микроцефалия, асимметрия лица, высокий и выступающий лоб, короткий бульбообразный нос, высокое нёбо, микроретрогнатия, низко посаженные ушные раковины, короткая шея, узкая и деформированная грудная клетка, крипторхизм, гипогонадизм. Характерны пороки сердечно-сосудистой системы и почек. Нередко развиваются бронхиальная астма и дерматозы.
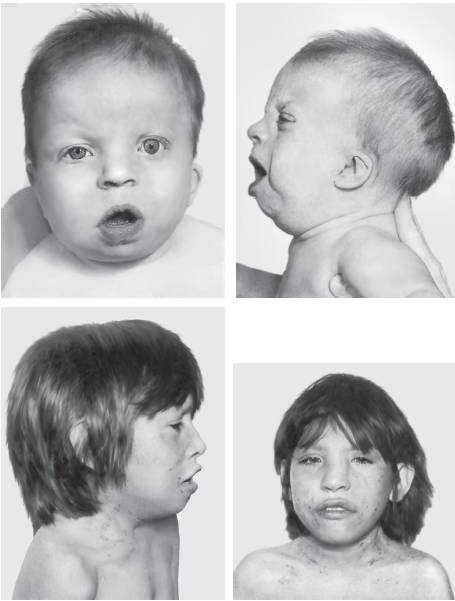
Рис. 22-18. Фенотип пациентов с мозаичной формой трисомии 14 (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
Анеуплоидии по половым хромосомам, как правило, характеризуются более мягкими клиническими симптомами по сравнению с дисбалансом числа аутосом.
У человека они представлены моносомией по хромосоме Х и различными вариантами полисомий по половым хромосомам.
Синдром Шерешевского-Тернера обусловлен моносомией по хромосоме Х. Это единственный вариант моносомии, совместимый с живорождением и постнатальным развитием организма. Кроме моносомии, этот синдром может развиваться при делециях длинного и короткого плеч хромосомы Х, изохромосомах и кольцевых хромосомах Х. В большинстве случаев (80-85%) единственная хромосома X имеет материнское происхождение. Распространены мозаичные формы заболевания с присутствием в организме клеток с нормальным хромосомным набором. Популяционная частота синдрома составляет 1:3000-5000 новорожденных. Клинические признаки заболевания (рис. 22-19): нанизм, крыловидные кожные складки на шее, короткая шея, бочкообразная грудная клетка, вальгусная девиация коленных и локтевых суставов, снижение зрения и слуха, отсутствие вторичных половых признаков. У больных отмечают первичную аменорею и бесплодие. Часто регистрируют врожденные пороки сердца и почек. Интеллектуальное развитие обычно соответствует норме.
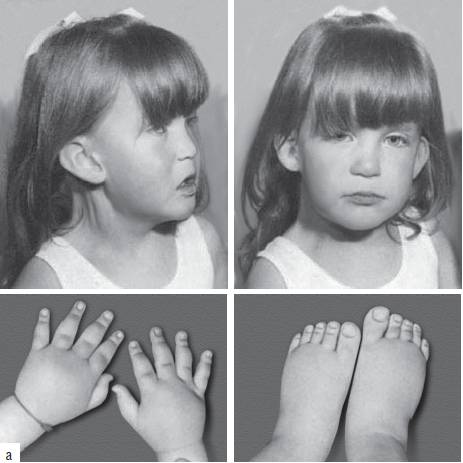
Рис. 22-19. Фенотипические особенности у пациентов с синдромом Шерешевского-Тернера в разном возрасте: a - 3 года (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.). Продолжение - на с. 577
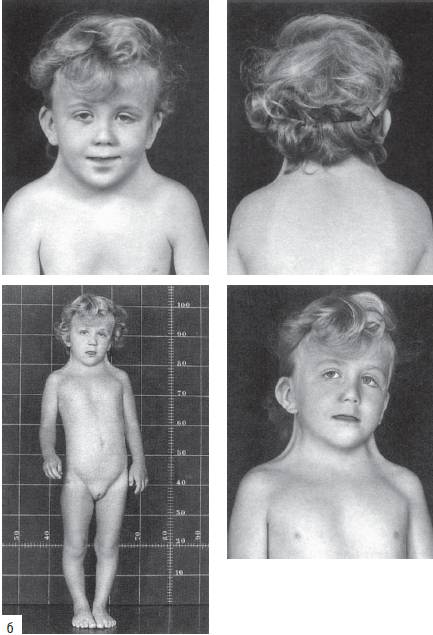
Рис. 22-19. Продолжение. Фенотипические особенности у пациентов с синдромом Шерешевского-Тернера в разном возрасте: б - 5 лет. Окончание - на с. 578
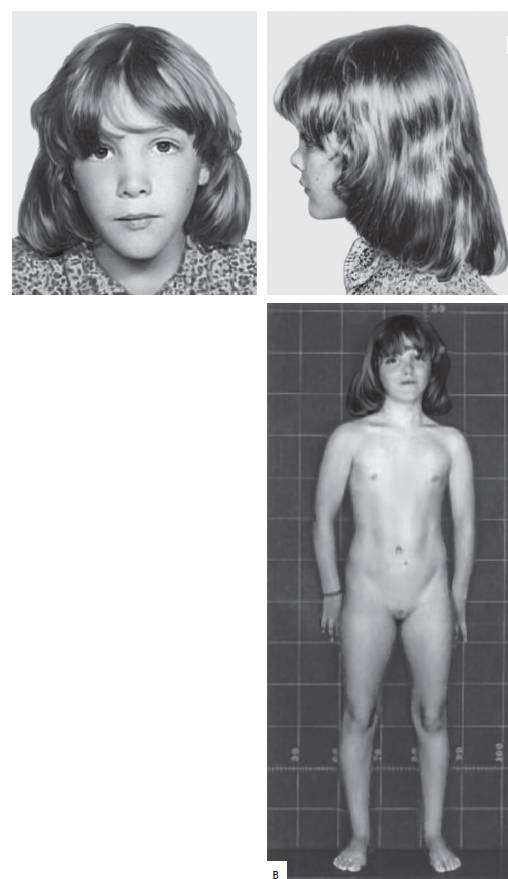
Рис. 22-19. Окончание. Фенотипические особенности у пациентов с синдромом Шерешевского-Тернера в разном возрасте: в - 12 лет.
Синдром трипло-Х формируется при носительстве кариотипа 47,ХХХ. Частота заболевания составляет один случай на 1000 новорожденных девочек. Как правило, женщины с таким хромосомным набором в полной или мозаичной форме имеют нормальное физическое и интеллектуальное развитие, что в значительной степени обусловлено инактивацией двух дополнительных хромосом Х. У женщин могут отсутствовать отклонения полового развития, но существует повышенный риск возникновения спонтанных абортов вследствие формирования анеуплоидных гамет. Лишь у некоторых пациенток отмечают нарушения репродуктивной функции в виде вторичной аменореи, дисменореи и ранней менопаузы.
С дальнейшим увеличением числа хромосом Х в кариотипе отклонения от нормы нарастают (рис. 22-20). У женщин с тетра- и пентасомией по хромосоме Х присутствуют черепно-лицевые дисморфии, аномалии зубов, скелета и половых органов. Способность к деторождению может сохраняться, но вследствие формирования анеуплоидных гамет возникает повышенный риск рождения детей с нарушениями числа хромосом Х.
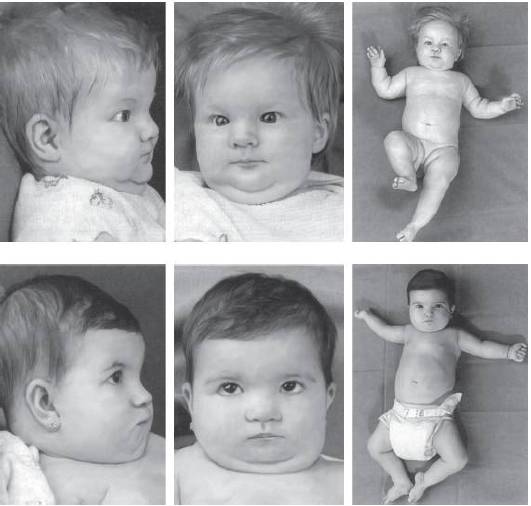
Рис. 22-20. Фенотип пациентов с тетрасомией (верхний ряд) и пентасомией (нижний ряд) по хромосоме X (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
Синдром Клайнфелтера объединяет присутствие в кариотипе не менее двух хромосом Х и не менее одной хромосомы Y. Цитогенетические формы представлены следующими вариантами: 47,XXY; 48,XXYY; 48,XXXY и 49,XXXXY. Наиболее распространенным считают кариотип 47,XXY, обнаруживаемый с частотой один случай на 1000 новорожденных мальчиков. Особенности клинической картины заболевания во многом связаны с появлением дополнительной хромосомы Х в кариотипе организма мужского пола. Такой дисбаланс манифестирует в период полового созревания и выражается в недоразвитии половых органов (гипогонадизм и гипогенитализм, дегенерация герминативного эпителия, гиалиноз семенных канатиков) и отсутствии вторичных половых признаков (рис. 22-21). Для больных с синдромом Клайнфелтера характерна азооспермия или олигоспермия. Из других клинических признаков необходимо отметить высокий рост, телосложение по женскому типу, гинекомастию, слабое оволосение лица, подмышечных впадин и лобка. Интеллект, как правило, снижен.
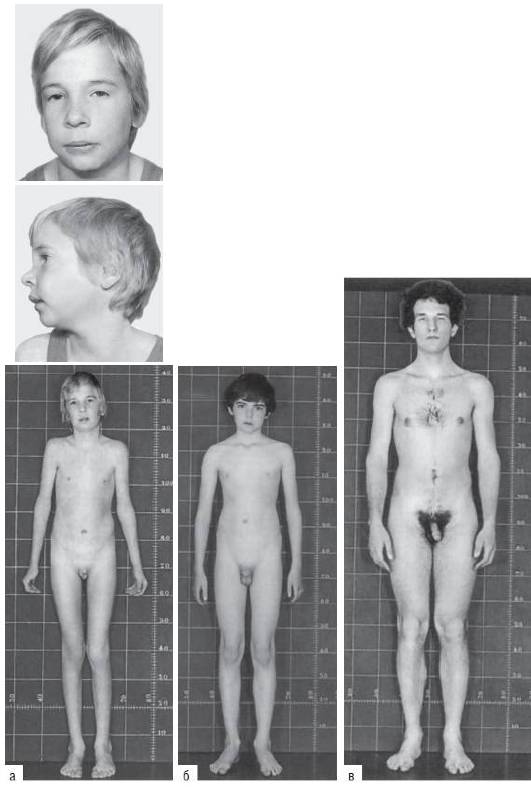
Рис. 22-21. Фенотип пациентов с синдромом Клайнфельтера в возрасте 6 (a), 13 (б) и 19 (в) лет (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
Синдром дисомии по хромосоме Y (47,XYY) регистрируют с частотой один случай на 1000 новорожденных мальчиков. Большинство носителей такого хромосомного набора имеют незначительные отклонения от нормального физического и интеллектуального развития (рис. 22-22). Обычно это индивидуумы с высоким ростом. Заметных нарушений полового развития и репродуктивной функции нет. У пациентов отмечают дефицит внимания, гиперреактивность и импульсивность.
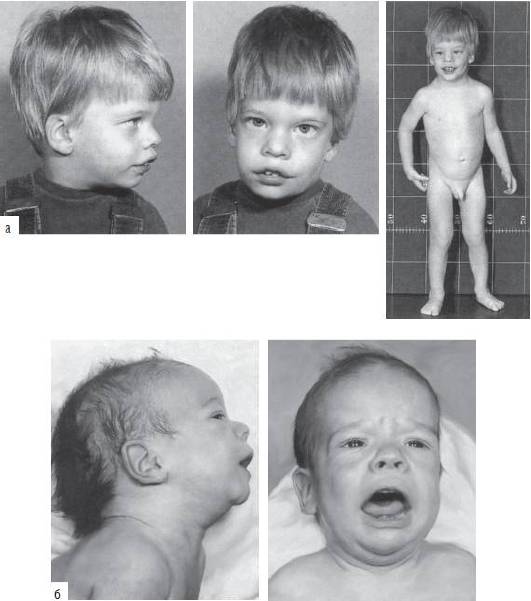
Рис. 22-22. Фенотип пациента с дисомией по Y-хромосоме (кариотип 47,XYY) в возрасте: а - 1 год и 10 мес; б - 2 мес (Schinzel A. Catalogue of Unbalanced Chromosome Aberrati ons in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.).
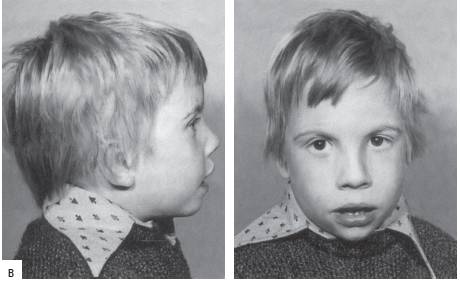
Рис. 22-22. Окончание. Фенотип пациента с дисомией по Y-хромосоме (кариотип 47,XYY) в возрасте: в - 5 лет.
Хромосомные болезни, обусловленные сегментными анеуплоидиями
Это сравнительно большая группа заболеваний, клиническая картина которых обусловлена структурными перестройками хромосом, приводящими к избытку (частичная трисомия) или недостатку (частичная моносомия) материала отдельных районов хромосом. Такие аберрации возникают de novo вследствие делеций или дупликаций тех или иных хромосомных сегментов в гаметогенезе или на ранних стадиях эмбриогенеза. Кроме того, несбалансированность по числу копий того или иного хромосомного региона может формироваться в результате наследования сбалансированных хромосомных перестроек (например, транслокаций) от родителей. Среди множества возможных структурных аберраций значимыми в отношении возникновения определенного хромосомного синдрома обычно считают только те, которые приводят к формированию сходной клинической картины у большинства пациентов.
Сегментные анеуплоидии, так же как и анеуплоидии по целым хромосомам, вызывают резкие отклонения в развитии. В большинстве случаев они не повторяют клиническую картину анеуплоидий по целым хромосомам. В связи с этим заболевания, обусловленные сегментными анеуплоидиями, часто выделяют в самостоятельные нозологические формы. Частичное совпадение клинических фенотипов у пациентов с полными и сегментными анеуплоидиями возможно в том случае, если последние затрагивают критические для развития хромосомного синдрома регионы хромосом (например, при болезни Дауна). Заболеваниям, обусловленным сегментными анеуплоидиями, свойственны общие клинические признаки всех хромосомных болезней: нарушения пре- и постнатального онтогенеза, врожденные пороки развития, умственная отсталость и сокращенная продолжительность жизни. На особенности клинического течения сегментной анеуплоидии могут влиять размер вовлеченного участка хромосомы и локализация точек разрыва, что определяет состав затрагиваемых перестройкой генов. Тонкая детализация структуры сегментных анеуплоидий в настоящее время стала возможной благодаря применению высокоразрешающих методов молекулярноцитогенетического и молекулярно-генетического исследования (микрочипов, секвенирования, микросателлитного анализа), позволяющих приблизиться к пониманию фенокариотипических корреляций, а в некоторых случаях дифференцировать новые хромосомные синдромы. Ниже будет приведена краткая характеристика некоторых наиболее распространенных синдромов, обусловленных сегментными анеуплоидиями хромосом.
Синдром Вольфа-Хиршхорна обусловлен делецией дистального региона короткого плеча хромосомы 4 (4p16.3). Частота синдрома варьирует в диапазоне 1:20 000-50 000 новорожденных, при этом среди девочек заболевание регистрируют в 2 раза чаще, чем среди мальчиков. Большинство случаев возникает de novo вследствие делеций, около 10% обусловлено наследственными формами. Основные клинические признаки: задержка пре- и постнатального роста, нарушения развития разной степени выраженности и низкая масса тела при рождении. Характерные черты лица и черепа (рис. 22-23): высокий лоб, микроцефалия, высокое надпереносье, клювовидный нос, гипертелоризм, выступающие глаза, короткий фильтр, микрогнатия, маленький рот с опущенными уголками рта, крупные оттопыренные уши. Часто обнаруживают расщелины губы и нёба. У мальчиков отмечают гипоспадию и крипторхизм. Характерны пороки развития сердечно-сосудистой системы и поликистоз почек. Один из ведущих клинических признаков - задержка психомоторного развития.
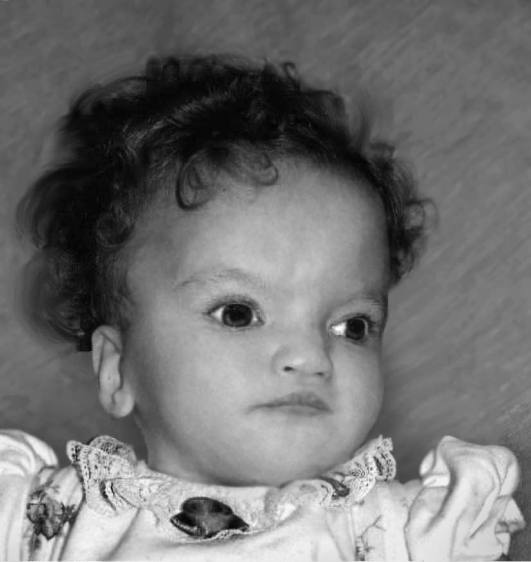
Рис. 22-23. Фенотип пациента с синдромом Вольфа-Хиршхорна
Синдром кошачьего крика - первое хромосомное заболевание, для которого в 1963 г. Ж. Лежен и соавт. продемонстрировали этиологическую роль структурного нарушения хромосом. Заболевание обусловлено делецией короткого плеча хромосомы 5. Возможна чрезвычайно небольшая делеция, затрагивающая критический сегмент 5p15.2, или отсутствие практически целого короткого плеча хромосомы. Популяционная частота синдрома составляет один случай на 20 000- 50 000 новорожденных. Среди пациентов с тяжелой умственной отсталостью (IQ <20) частота синдрома кошачьего крика составляет около 1%. Клинические признаки заболевания обусловлены гаплонедостаточностью большого числа генов, возникающей вследствие делеций хромосомного региона (рис. 22-24). Среди клинически значимых признаков чаще всего обнаруживают необычный крик или плач, напоминающий мяуканье кошки, микроцефалию, антимонголоидный разрез глаз, лунообразное лицо, гипертелоризм, широкую переносицу и низко посаженные деформированные ушные раковины (рис. 22-25). На ладонях присутствует поперечная складка. Кроме того, характерна клинодактилия или синдактилия, а также тяжелая умственная отсталость. Продолжительность жизни зависит от тяжести пороков внутренних органов. Больные погибают в основном в первые годы жизни.
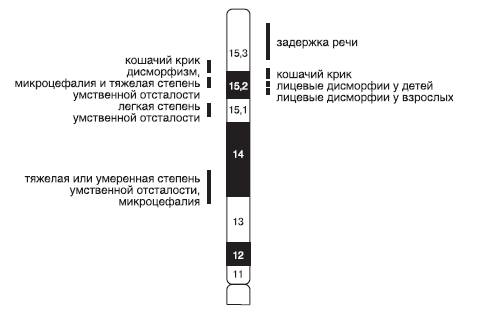
Рис. 22-24. «Фенотипическая карта» короткого плеча хромосомы 5 (Mainardi P.C. Cri du Chat syndrome // Orphanet J. Rare Dis. - 2006. - Vol. I. - P. 33)

Рис. 22-25. Пациенты с синдромом кошачьего крика в возрасте: а - 8 мес; б - 2 лет; в - 4 лет; г - 9,5 лет. (Mainardi P.C. Cri du Chat syndrome // Orphanet J. Rare Dis. - 2006. - Vol. I. - P. 33)
Синдром трисомии 9p - одна из самых распространенных частичных трисомий. Впервые описан в 1970 г. Возникновение заболевания может быть связано с дупликациями, изохромосомами по короткому плечу хромосомы 9 и несбалансированными транслокациями. Основные признаки заболевания (рис. 22-26): антимонголоидный разрез глаз, гипертелоризм, микробрахицефалия, энофтальм, широкий и округлый кончик носа, выступающие верхняя губа и верхняя челюсть, низко посаженные ушные раковины с аномальным противозавитком и противокозелком, узкий слуховой канал, короткая шея с низкой линией роста волос и поперечная ладонная складка. Рентгенографически обнаруживают задержку костного возраста. У пациентов отмечается олигофрения. Прогноз жизни благоприятный; больные доживают до пожилого возраста.
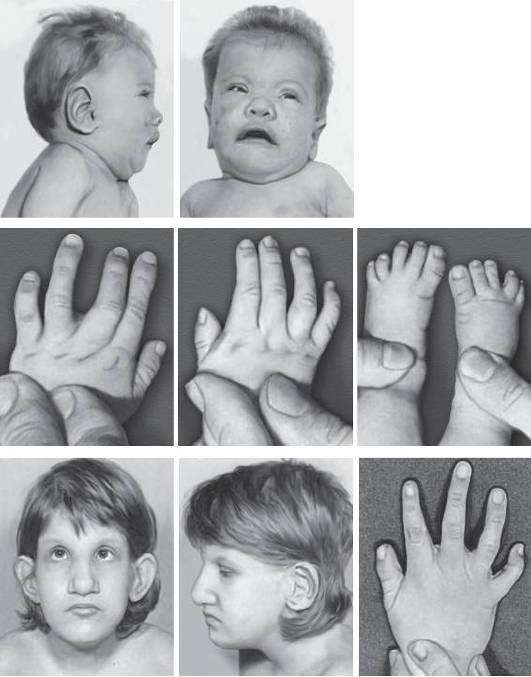
Рис. 22-26. Фенотипические особенности пациентов с дупликацией короткого плеча хромосомы 9 в возрасте 4 года (верхний и средний ряд) и 12 лет 4 мес (нижний ряд) (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
Синдром моносомии 9p (синдром Альфи) впервые описан Альфи и соавт. в 1973 г. Он возникает вследствие частичных делеций короткого плеча хромосомы 9, изохромосом по длинному плечу и несбалансированных транслокаций. Критический регион - сегмент 9p22. Основные клинические признаки (рис. 22-27) заболевания: тригоноцефалия, резко выступающий лоб, монголоидный разрез глаз, эпикант, экзофтальм, гипертелоризм, уплощенная широкая переносица, маленький рот с большой верхней губой и высокое нёбо. Ушные раковины со сглаженным завитком не имеют мочки или она недоразвита. Характерны короткая шея и гипертелоризм сосков; длинные пальцы рук и ног. У девочек отмечают гипоплазию больших и малых половых губ, у мальчиков - гипоплазию мошонки и полового члена. Умственная отсталость достигает стадии имбецильности или дебильности. Прогноз жизни благоприятный.
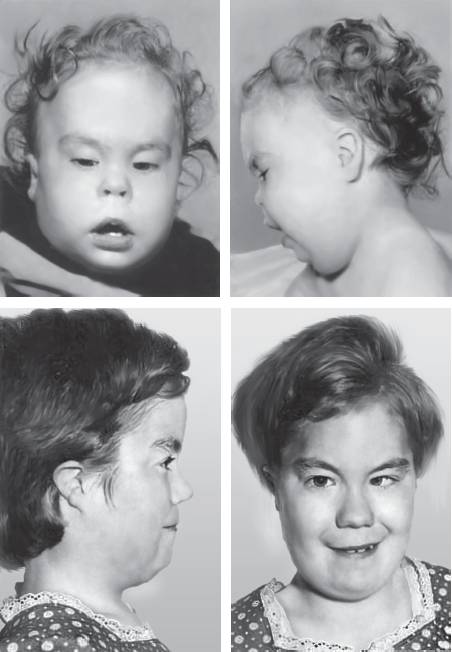
Рис. 22-27. Фенотип пациентов с делецией 9pter-p21 в возрасте 10 мес и 13,5 лет. [Верхний ряд: Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.; нижний ряд: Rutten F.J., Hustinx T.W.J., Dunk-Tillemans A.A.W. et al. A case of partial 9p monosomy with some unusual features // Ann. Genet. (Paris). - 1978. - Vol. 21. - P. 51-55]
Синдром моносомии 13q (синдром Орбели) формируется при наличии в кариотипе кольцевой хромосомы 13 или делеций. Критический регион - сегмент 13q14. Для детей с этим заболеванием характерна низкая масса тела при рождении. Основные признаки синдрома: микроцефалия, асимметрия лица, широкая выступающая переносица, отсутствие носовой вырезки, антимонголоидный разрез глаз и высокое нёбо (рис. 22-28). Отмечают поражения опорно-двигательного аппарата: короткую шею, гипоили аплазию I пальца кисти и пяточной кости, а также клинодактилию. Характерны пороки сердца, почек, головного мозга, заращение прямой кишки и заднепроходного отверстия. Для больных характерна глубокая олигофрения. Большинство пациентов погибают в течение первого года жизни.
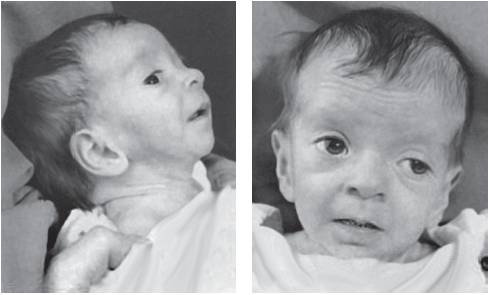
Рис. 22-28. Пациент с делецией 13q13-31 в возрасте 2 мес. (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
Синдром трисомии 14q возникает вследствие несбалансированных транслокаций, дупликаций или изохромосом по длинному плечу хромосомы 14. Основные признаки заболевания: микроцефалия, луковицеобразный нос, тонкая верхняя губа, микростомия, опущенные углы рта, низко расположенные ушные раковины, высокое арковидное нёбо или расщелина нёба, короткая шея, клинодактилия, косолапость, пороки сердечно-сосудистой системы, отставание в физическом и психомоторном развитии.
Синдром моносомии 18p (синдром де Груши). Впервые описан в 1963 г. Основные клинические признаки заболевания: умственная отсталость, задержка роста, а также аномалии развития лица, черепа (включая характерное круглое лицо, диспластичные уши, широкий рот, нарушения строения зубов), конечностей, гениталий, сердца и головного мозга (рис. 22-29, 22-30). Отмечено, что характерное для неонатального периода круглое лицо в детстве может становиться вытянутым.
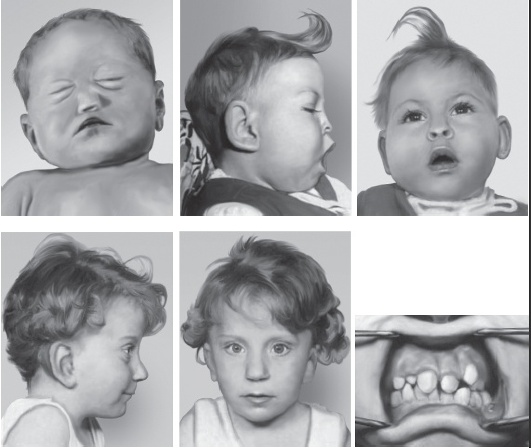
Рис. 22-29. Фенотипические особенности пациентов с моносомией 18p в возрасте 10 мес (верхний ряд) и 4 лет (нижний ряд) (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
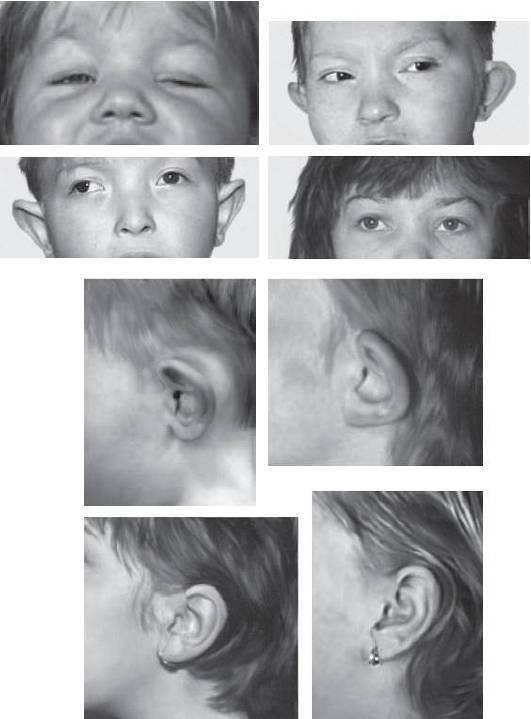
Рис. 22-30. Лицевые аномалии у пациентов с синдромом моносомии 18p (Turleau C. Monosomy 18p // Orphanet J. Rare Dis. - 2008. - Vol. 3. - P. 4). Окончание - на с. 590
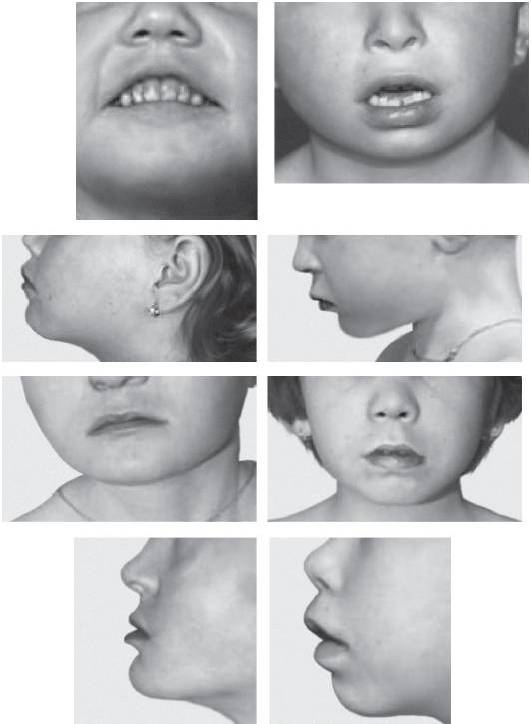
Рис. 22-30. Окончание
Синдром моносомии 18q возникает вследствие делеций длинного плеча хромосомы 18. Типичные признаки заболевания: микроцефалия, гипоплазия средней части лица, глубоко посаженные глаза, «рот карпа», плоский или вогнутый профиль лица, высокое нёбо или его расщелина, деформированные ушные раковины с атрезией или сужением слуховых проходов и аномалии конечностей (конические пальцы, клинодактилия V пальца, вальгусная деформация тазобедренного сустава). Характерны патологические изменения органа зрения, включающие нистагм, страбизм, эпикант и атрофию зрительных нервов. У мальчиков отмечают крипторхизм, гипоплазию полового члена и мошонки. У девочек обнаруживают гипоплазию половых губ. Регистрируют пороки развития сердца и почек. Умственная отсталость, как правило, достигает глубокой степени.
Синдром моносомии 21q обусловлен делецией длинного плеча хромосомы, а также кольцевой хромосомой 21 (рис. 22-31). Впервые описан Леженом в 1964 г. Основные признаки заболевания: низкая масса тела при рождении, микроцефалия, антимонголоидный разрез глаз, микрофтальмия, колобома радужки, катаракта, эпикант, выступающая широкая переносица, большие, низко посаженные уши с расширенным наружным слуховым каналом, микрогнатия, клинодактилия, сколиоз, мышечная гипертензия, гипоспадия, крипторхизм и косолапость. Характерны пороки сердца и почек (агенезия, гидронефроз, удвоение почечных лоханок), а также задержка психомоторного развития.
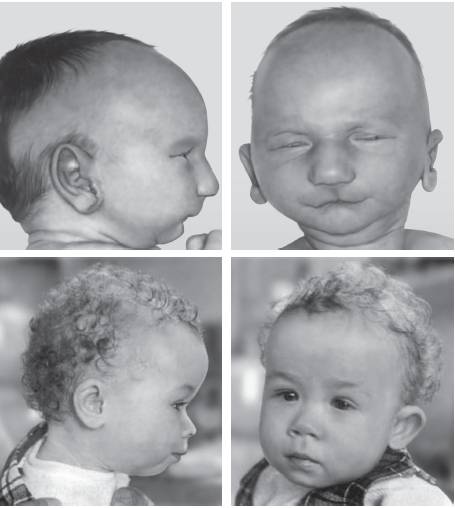
Рис. 22-31. Новорожденный с кольцевой хромосомой 21 (верхний ряд) и пациент в возрасте 7 мес (нижний ряд) (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p. Фотография новорожденного использована из работы: Richer C.L., Fitch N., Sitahal S. et al. Analysis of banding patterns in a case of ring chromosome 21 // Am. J. Med. Genet. - 1981. - Vol. 10. - P. 323-331. Фотография пациента с кольцевой хромосомой 21 приведена из статьи: Shibata K., Waldenmaier C., Hirsch W. A child with a 21-ring chromosome 45,XX,21-46,XX,21r investigated with the banding technique // Hum. Genet. - 1973. - Vol. 18. - P. 315-319)
Синдром кольцевой хромосомы 18 впервые описан в 1962 г. Основные клинические признаки заболевания: микроцефалия, гипертелоризм, страбизм, птоз, нистагм, «рот карпа», тонкая верхняя губа и высокое нёбо (часто с расщелиной). Умственная отсталость, как правило, достигает глубокой степени тяжести (рис. 22-32, 22-33).
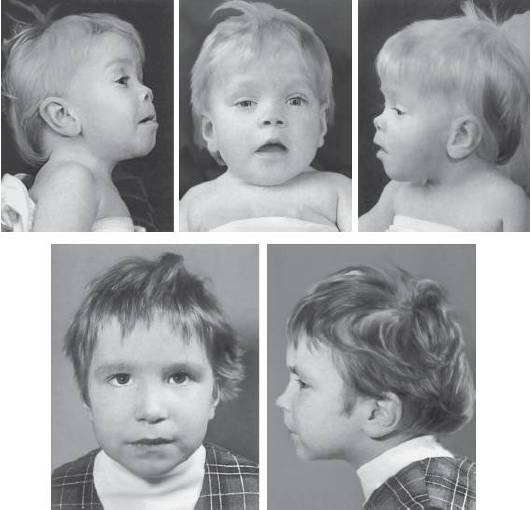
Рис. 22-32. Фенотип пациентов с кольцевой хромосомой 18 в возрасте 13 мес (верхний ряд) и 6 лет (нижний ряд) (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
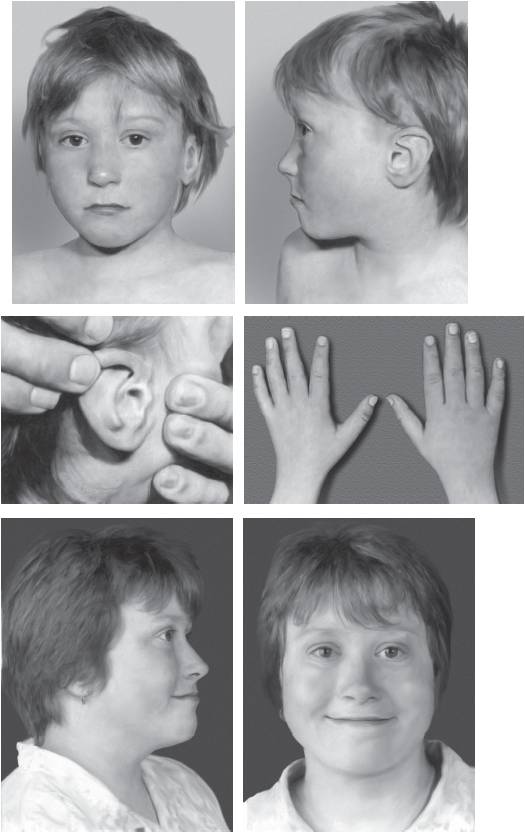
Рис. 22-33. Фенотип пациентки с кольцевой хромосомой 18 в возрасте 6 и 17 лет (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
Синдром кольцевой хромосомы 22 как самостоятельная нозологическая форма впервые описан в 1972 г. Развитие заболевания может быть обусловлено не только собственно кольцевой хромосомой, но и делецией длинного плеча хромосомы 22. В 90% случаев подобные мутации возникают de novo. Основные клинические признаки синдрома: микроцефалия, птоз, эпикант, гипертелоризм, крупные выступающие глаза, расщелина языка и нёба, дисплазия тазобедренных суставов, клинодактилия и синдактилия (рис. 22-34). Умственная отсталость - от легкой степени до выраженной олигофрении. Для больных типичны легкая возбудимость, частая смена настроения, нарушения координации и глубокое недоразвитие речи, вплоть до ее отсутствия.
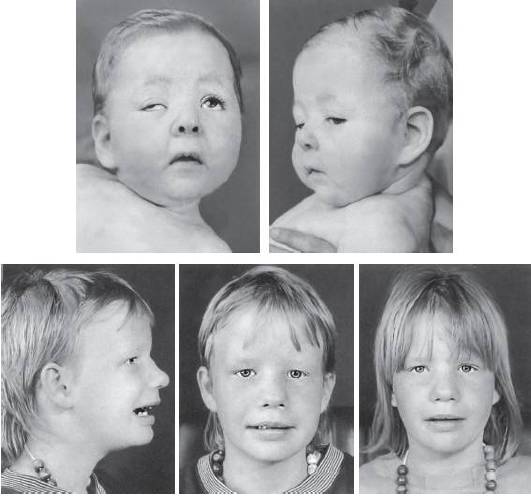
Рис. 22-34. Фенотип пациентов с кольцевой хромосомой 22 в возрасте 8 мес (верхний ряд) и 11 лет (нижний ряд) (Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
Синдромы, обусловленные микроструктурными аберрациями хромосом
В эту группу входят синдромы, обусловленные незначительными, размером до 5 млн пар нуклеотидов, делециями или дупликациями определенных участков хромосом. Соответственно их называют микроделеционными и микродупликационными синдромами. Многие из этих синдромов первоначально были описаны как доминантные заболевания, обусловленные точечными мутациями генов, но позднее с помощью методов молекулярно-цитогенетического анализа была установлена их истинная этиология. С использованием сравнительной геномной гибридизации на микрочипах стало возможным обнаружение делеций и дупликаций хромосом протяженностью до одного гена с прилегающими областями, что позволило не только существенно расширить список микроделеционных и микродупликационных синдромов, но и приблизиться к пониманию генофенотипических корреляций у пациентов с микроструктурными аберрациями хромосом.
Один из механизмов формирования микроделеций или микродупликаций - неаллельная рекомбинация гомологичных хромосом в регионах локализации сегментных дупликаций генома. Это особый класс низкокопийных повторов ДНК, представленный крупными блоками размером до 100 тыс. пар нуклеотидов с высокой степенью (>95%) идентичности нуклеотидных последовательностей. Часто сегментные дупликации кластеризуются в прицентромерных и субтеломерных районах хромосом. В геноме человека такие блоки отмечены в хромосомах 7, 15, 17, 22 и Х (рис. 22-35, см. цв. вклейку). Неравный кроссинговер между блоками сегментных дупликаций приводит к возникновению микроделеций и микродупликаций, которые в зависимости от размера обусловливают формирование хромосомного или моногенного заболевания.
Очевидно, что в основе развития микроделеционных и микродупликационных синдромов лежат изменения дозы генов в участке хромосомы, затронутом перестройкой. Пока не установлено, что именно составляет основу формирования большинства таких синдромов - отсутствие конкретного структурного гена или более протяженного участка, содержащего несколько генов. Болезни, которые возникают вследствие микроделеций участка хромосомы, содержащего несколько генных локусов, предложено называть смежными генными синдромами. Для формирования клинической картины этой группы заболеваний принципиально важным считают отсутствие продукта нескольких генов, затрагиваемых микроделецией. По своей этиологии смежные генные синдромы находятся на границе между менделирующими моногенными заболеваниями и хромосомными болезнями (рис. 22-36). Типичный пример такого заболевания - синдром Прадера-Вилли, возникающий вследствие микроделеции размером 4 млн пар нуклеотидов в регионе q11-q13 на хромосоме 15 отцовского происхождения. Микроделеция при синдроме Прадера- Вилли затрагивает 12 импринтированных генов (SNRPN, NDN, MAGEL2 и ряд других), которые в норме экспрессируются только с отцовской хромосомы. Точковые мутации в каждом из них возможны, но для развития заболевания критическим считают отсутствие экспрессии всего набора импринтированных генов.
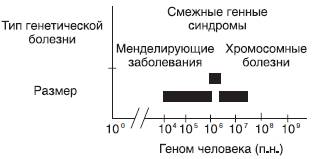
Рис. 22-36. Размеры геномных перестроек при различных типах генетических болезней (Stankiewicz P., Lupski J.R. Genome architecture, rearrangements and genomic disorders // Trends Genet. - 2002. - Vol. 18. - P. 74-82)
Интересен тот факт, что микроделеции и микродупликации одного и того же хромосомного региона, будучи по отношению друг к другу реципрокными продуктами неаллельной гомологичной рекомбинации, часто ведут к разным хромосомным синдромам, которые тем не менее могут иметь некоторые общие клинические признаки. Примеры таких заболеваний: синдромы Смита-Магениса (del17p11.2) и Пото ки-Лупски (dup17p11.2); синдромы Миллера-Дикера (del17p13.3) и дупликации 17p13.3.
Пока остается неясным, как влияет состояние локуса в гомологичной хромосоме на клиническое течение микроделеционных синдромов. По-видимому, происхождение клинических признаков разных синдромов различно. Патологический процесс при некоторых из них развертывается через инактивацию опухолесупрессорных генов (опухоль Вильмса), а клиническая картина других синдромов обусловлена не только делециями, но и явлениями хромосомного импринтинга и однородительских дисомий (синдромы Прадера-Вилли, Ангельмана, Видеманна-Беквита). В табл. 22-3 приведены примеры некоторых синдромов, обусловленных микроделециями или микродупликациями небольших фрагментов хромосом. Большинство из них регистрируют крайне редко (1:50 000-100 000 новорожденных). Как правило, они имеют отчетливую клиническую картину и диагноз можно поставить по совокупности симптомов. Тем не менее в связи с прогнозом здоровья будущих детей в семье, в том числе у родственников родителей пробанда, необходимо провести высокоразрешающее цитогенетическое исследование у пробанда и его родителей.
Таблица 22-3. Характеристика микроделеционных и микродупликационных синдромов
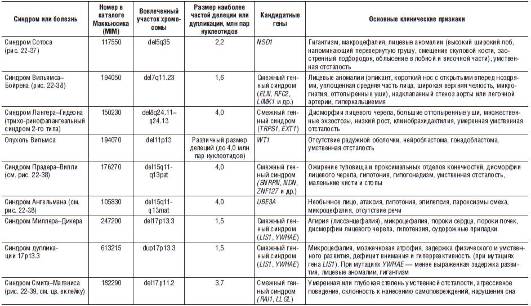
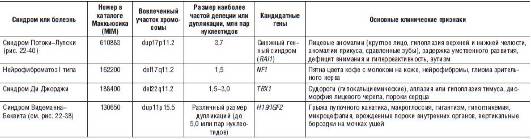
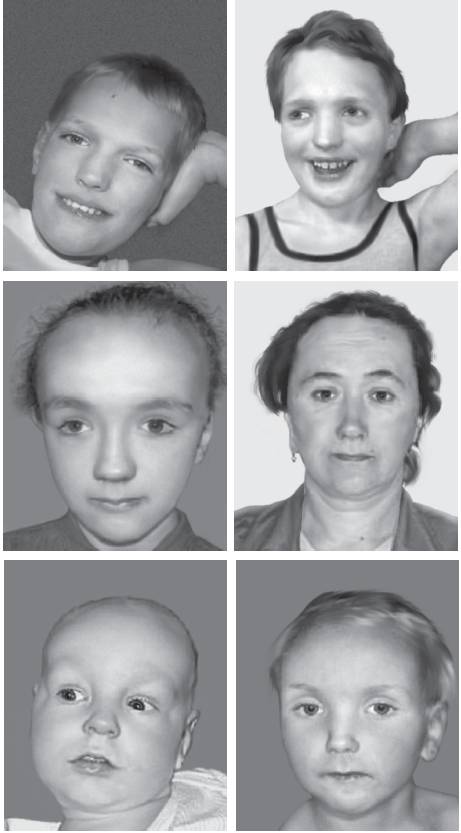
Рис. 22-37. Лицевые аномалии у пациентов с синдромом Сотоса (Türkmen S., Gillessen-Kaesbach G., Meinecke P. et al. Mutations in NSD1 are responsible for Sotos syndrome, but are not a frequent finding in other overgrowth phenotypes // Eur. J. Hum. Genet. - 2003. - Vol. 11. - P. 858-865).
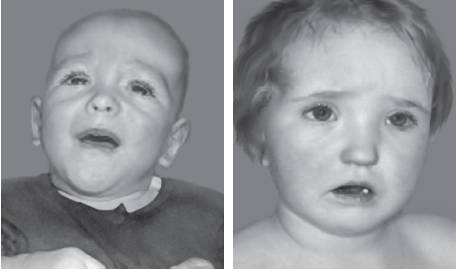
Рис. 22-37. Окончание
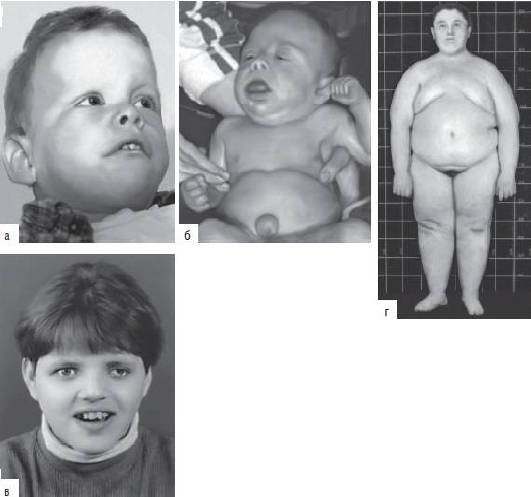
Рис. 22-38. Фенотип пациентов с микроделеционными синдромами: a - синдром Вильямса-Бойрена (Heller R., Rauch A., Luttgen S. et al. Partial deletion of the critical 1.5 Mb interval in Williams-Beuren syndrome. Online mutation report // J. Med. Genet. - 2003. - Vol. 40. - P. e99 doi:10.1136/jmg.40.8.e99); б - синдром Видеманна-Беквита (Weksberg R., Shuman C., Beckwith J.B. Beckwith-Wiedemann syndrome // Eur. J. Hum. Genet. - 2010. - Vol. 18. - P. 8-14); в - синдром Ангельмана; г - синдром Прадера-Вилли (в , г - Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.)
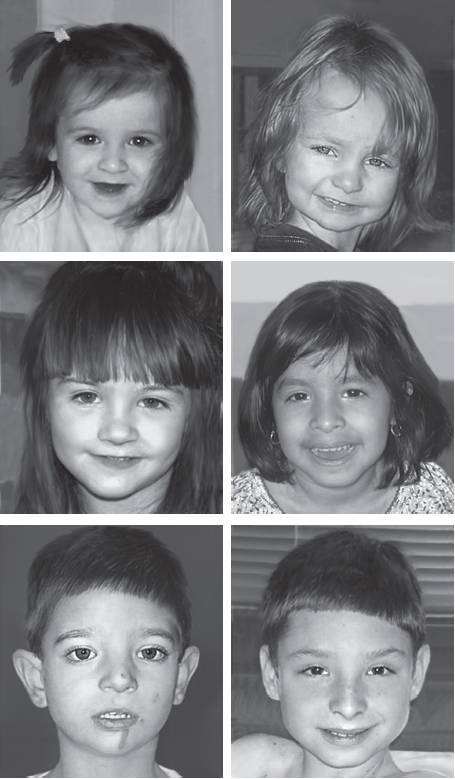
Рис. 22-40. Фенотип пациентов с синдромом Потоки-Лупски [Potocki L., Bi W., Treadwell-Deering D., et al. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype // Am. J. Hum. Genet. - 2007. - Vol. 80. - P. 633-649]. Окончание - на с. 601
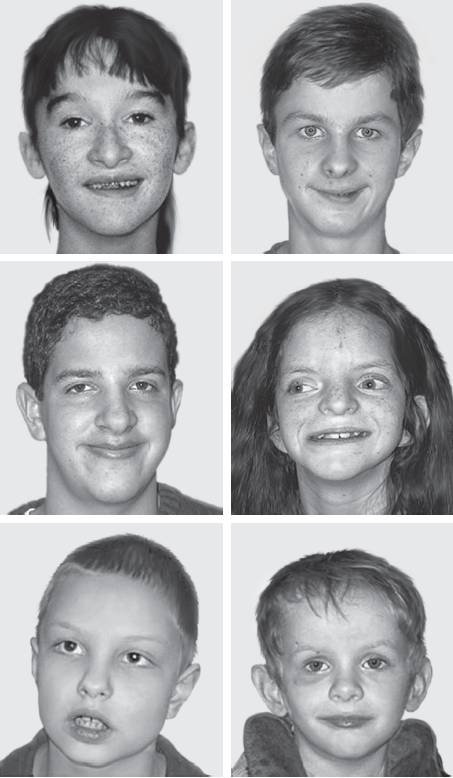
Рис. 22-40. Окончание
В отдельную группу микроделеционных синдромов выделяют хромосомные заболевания, возникающие вследствие утраты субтеломерных участков хромосом. Диагностика таких перестроек с помощью стандартного метафазного анализа G-окрашенных препаратов достаточно затруднительна вследствие ограниченных разрешающих возможностей, накладываемых световой микроскопией. Внедрение в практику диагностики хромосомных болезней FISH-метода с использованием уникальных ДНК-зондов, комплементарных субтеломерным районам хромосом, а также применение микрочиповых технологий позволило уточнить цитогенетическую этиологию ряда патологических состояний, характеризующихся умственной отсталостью различной степени выраженности и множественными врожденными пороками развития. В настоящее время известны хромосомные синдромы, обусловленные аномалиями субтеломерных регионов практически каждой хромосомы в кариотипе человека (de Vries et al., 2003; рис. 22-41). Ниже перечислены некоторые из них.
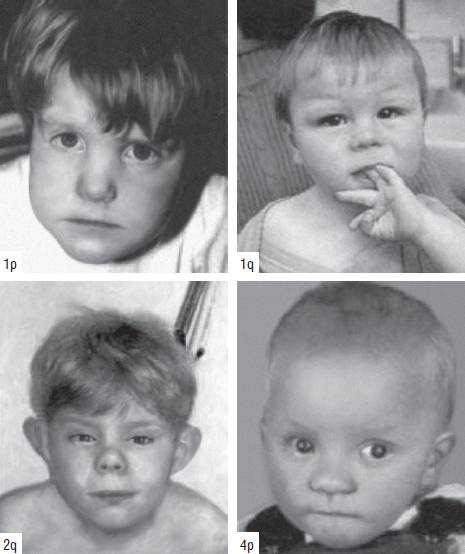
Рис. 22-41. Фенотипы пациентов с микроделециями в субтеломерных регионах хромосом (De Vries B.B.A., Winter R., Schinzel A., van Ravenswaaij-Arts. Telomeres: a diagnosis at the end of the chromosomes // J. Med. Genet. - 2003. - Vol. 40. - P. 385-398). Окончание - на с. 603
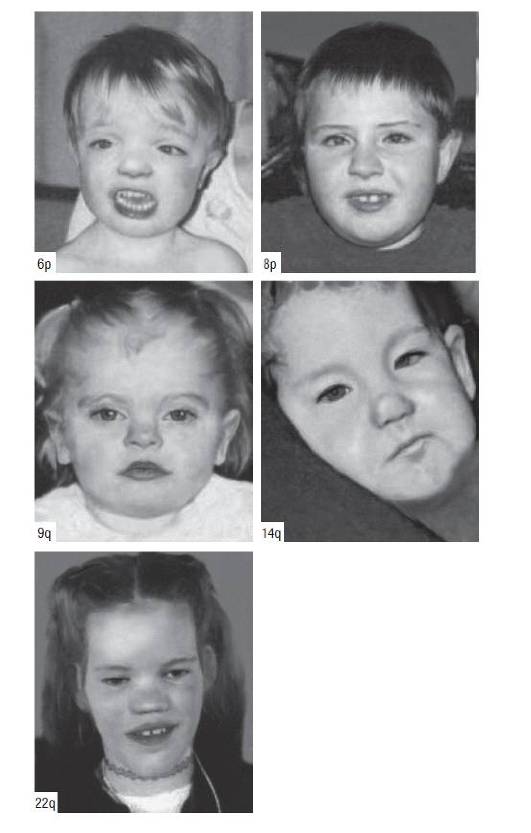
Рис. 22-41. Окончание
Синдром частичной моносомии 1p36 является самым распространенным микроделеционным синдромом. Его частота составляет 1:5000 новорожденных. 95% делеций возникают de novo, из них 60% происходит на хромосомах материнского происхождения, а 40% - на хромосомах, наследованных от отца. Известны терминальные и интерстициальные формы делеций. Клиническими признаками синдрома являются: задержка развития, умственная отсталость от умеренной до сильной степени тяжести, нарушения слуха, эпилептические приступы, гипотония, пороки сердца и ряд дисморфических черт лица, среди которых выделяются уплощенная переносица, глубоко посаженные глаза, гипертелоризм, гипоплазия средней части лица, заостренный подбородок (рис. 22-42). Кандидатными генами для данного синдрома рассматриваются протоонкоген SKI, ген β-субъединицы калиевого канала KCNAB2, а также ген матричной металлопротеиназы MMP23, вовлеченные, как предполагают, в патогенез лицевых дисморфических признаков, эпилепсии и краниосиностоза соответственно.

Рис. 22-42. Лицевые аномалии у пациентов с микроделецией 1p36: а - пациент в возрасте 13 лет и 7 мес; б - пациент в возрасте 6 лет и 5 мес; в и г - один и тот же пациент в возрасте 9 и 15 лет [Gajecka M., Mackay K.L., Shaffer L.G. Monosomy 1p36 deletion syndrome// Am. J. Med. Genet. Part С (Seminars in Medical Genetics). - 2007. - Vol. 145C. - P. 346-356]
Синдром Питта-Роджера-Данкса (MIM 194190) обусловлен микроделецией 4p16.3 протяженностью менее 3,5 млн пар нуклеотидов. По существу, клинически это более мягкий вариант синдрома Вольфа-Хиршхорна. Характеризуется отставанием массы тела от нормы, микроцефалией и умственной отсталостью.
Синдром Куррарино (MIM 176450) связан с микроделецией 7q36, приводящей к потере гомеобоксного гена HLXB9. Характерные клинические признаки - крестцовый дисгенез и аноректальная атрезия.
Синдром Джекобсена (MIM 147791), или синдром частичной моносомии 11q, обусловлен микроделециями субтеломерных сегментов q23, q24 и q25 длинного плеча хромосомы 11. Клинически манифестирует умеренной задержкой умственного развития и задержкой постнатального роста. Среди признаков заболевания отмечают тригоноцефалию, гипертелоризм, эпикант, птоз, короткий нос, длинный фильтр, ретрогнатию, аномалии конечностей, пороки сердца и тромбоцитопению.
Синдром α-талассемии, сцепленный с умственной отсталостью, или синдром ATR-16 (MIM 141750). Развитие этого смежного генного синдрома обусловлено микроделецией 16p13.3, ведущей к утрате α-глобиновых генов HBA1 и HBA2, а также гена SOX8, экспрессирующегося в головном мозге. С потерей последнего связывают формирование умственной отсталости. Основные клинические признаки синдрома: лицевые аномалии (гипертелоризм, скошенные вниз глазные щели, широкая уплощенная переносица), α-талассемия и умственная отсталость.
При микроделециях субтеломерных районов некоторых хромосом клиническая картина заболевания может напоминать или иметь менее выраженные признаки синдрома, связанного с потерей более протяженного участка хромосомы. Так, например (как было отмечено ранее), синдром Питта-Роджера-Данкса - более мягкий вариант синдрома Вольфа-Хиршхорна. Сходную ситуацию наблюдают при синдромах кошачьего крика (del5p), моносомии 9p, Миллера-Дикера (del17p13.3) и де Груши (del18q21, del18q23).
Современное развитие молекулярно-цитогенетических технологий, и прежде всего сравнительной геномной гибридизации на микрочипах (array CGH), создало предпосылки для идентификации тонких микроструктурных аберраций хромосом, обычно не обнаруживаемых при использовании рутинных методов цитогенетической диагностики. Серия работ по скринингу пациентов с умственной отсталостью или множественными врожденными пороками развития позволила описать ряд новых микроделеционных и микродупликационных синдромов (Slavotinek, 2008). Кратко остановимся на характеристике мутаций, вызывающих эти заболевания, а также на особенностях их клинической картины.
Синдром частичной моносомии 1q41-42 обусловлен микроделецией протяженностью 1,17 млн пар нуклеотидов, затрагивающей ген DISP1. Большинство случаев микроделеции возникает de novo. Заболевание характеризуется умеренной или тяжелой формой умственной отсталости. Пациенты имеют лицевые аномалии (крупное лицо, глубоко посаженные глаза, гипертелоризм, уплощенная переносица, вывернутые вперед ноздри, широкий кончик носа, полные губы), расщелину нёба, косолапость, диафрагмальную грыжу, короткие тонкие пальцы, кожную синдактилию и гипоплазию ногтевой пластины.
Синдром частичной моносомии 2p15-16.1 обусловлен микроделециями, варьирующими от 570 тыс. до 5,7 млн пар нуклеотидов. Минимальный критический регион на хромосоме 2 составляет всего 200 тыс. пар нуклеотидов. Кандидатный ген заболевания - VRK2. Клинические особенности (рис. 22-43): микроцефалия, телекант, птоз век, страбизм, высокая перемычка носа, крупные, низко посаженные уши, вывернутая верхняя губа, высокое нёбо, камптодактилия, аномалии почек, спазм мышц конечностей, гипоплазия зрительного нерва, гипоплазия мозжечка и ствола головного мозга, пахигирия и аутизм.
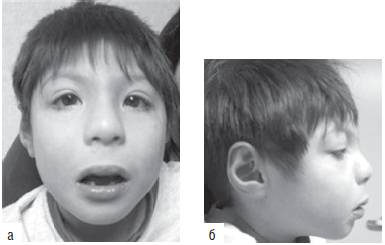
Рис. 22-43. Фенотип пациента с микроделецией 2p15-16.1 в возрасте 8 (a) и 13 лет (б) (Slavotinek A.M. Novel microdeletion syndromes detected by chromosome microarrays // Hum. Genet. - 2008. - Vol. 124. - P. 1-17.) Автор использовал иллюстрации из работы: Rajcan-Separovic E, Harvard C, Liu X. et al. Clinical and molecular cytogenetic characterization of a newly recognised microdeletion syndrome involving 2p15-16.1 // J. Med. Genet. - 2007. - Vol. 44. - P. 269-276 с разрешения издательств Macmillan Publishers Ltd и Nature Publishing Group
Синдром частичной моносомии 9q22.3 связан с микроделецией размером 6,5 млн пар нуклеотидов, затрагивающей гены GABA-BR2 и TGFBR1. Эта одна из наиболее частых мутаций, обнаруживаемых примерно у 4% пациентов с умственной отсталостью. Клинически сопровождается пре- и постнатальным увеличением роста, тригоноцефалией, страбизмом, маленьким ртом, низко посаженными ушами, тонкой верхней губой, короткой шеей, пупочной грыжей, расслаблением суставов, увеличением костного возраста, вентрикуломегалией и церебральной атрофией (рис. 22-44). У пациентов отмечают тяжелую степень умственной отсталости и гиперреактивность.
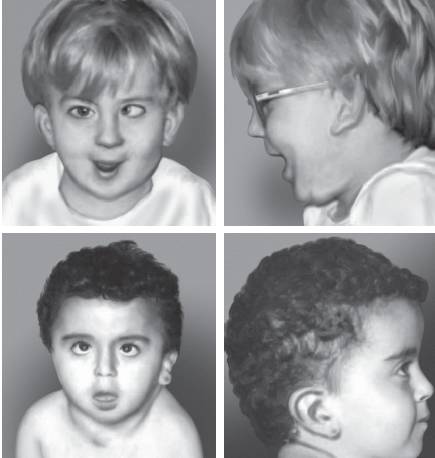
Рис. 22-44. Фенотип пациентов с микроделецией 9q22.3 (Slavotinek A.M. Novel microdeletion syndromes detected by chromosome microarrays // Hum. Genet. - 2008. - Vol. 124. - P. 1-17). Автор использовал иллюстрации из работы: Redeon R., Baujat G., Sanlaville D. et al. Interstitial 9q22.3 microdeletion: clinical and molecular characterisation of a newly recognized overgrowth syndrome // Eur. J. Hum. Genet. - 2006. - Vol. 14. - P. 759-767 с разрешения издательств Macmillan Publishers Ltd и Nature Publishing Group A.M. Novel microdeletion syndromes detected by chromosome microarrays // Hum. Genet. - 2008. - Vol. 124. - P. 1-17.) Автор использовал иллюстрации из работы: Rajcan-Separovic E, Harvard C, Liu X. et al. Clinical and molecular cytogenetic characterization of a newly recognised microdeletion syndrome involving 2p15-16.1 // J. Med. Genet. - 2007. - Vol. 44. - P. 269-276 с разрешения издательств Macmillan Publishers Ltd и Nature Publishing Group
Синдром частичной моносомии 15q13.3 обусловлен микроделецией размером 1,5 млн пар нуклеотидов, затрагивающей ген CHRNA7. Клинически манифестирует умственной отсталостью легкой или умеренной степени тяжести, аутизмом и гипотензией. Для пациентов характерны лицевые аномалии - гипертелоризм, выступающий фильтр и вывернутая утолщенная нижняя губа.
Синдром частичной моносомии 15q24 вызывают микроделеции минимальным размером 1,7 млн пар нуклеотидов, затрагивающие ген P450SCC. Клинические признаки заболевания - легкая или умеренная степень умственной отсталости, гиперреактивность, аутистические черты и счастливое выражение лица. Для пациентов характерны задержка пре- и постнатального роста, обусловленная дефицитом гормона роста, микроцефалия, высокий лоб, асимметрия лица, гипертелоризм, выступающий фильтр, аномалии ушной раковины, утолщенная нижняя губа, аномалии проксимальных отделов конечностей, крипторхизм и сколиоз.
Синдром частичной трисомии 17q21.31 является одним из самых распространенных синдромов, выявляемых с помощью матричной сравнительной геномной гибридизации. Его частота среди новорожденных оценивается в диапазоне 1:13000-1:20000, а среди пациентов с умственной отсталостью он может быть выявлен у 1% индивидуумов (Koolen et al., 2006). Клинически синдром характеризуется задержкой физического и умственного развития, выраженной неонатальной гипотонией, нарушением сосательного рефлекса и плохим вскармливанием, диспраксией, лицевыми аномалиями, среди которых отмечаются: вытянутое лицо, птоз, блефарофимоз, бульбообразный кончик носа, крупные и низко посаженные уши (рис. 22-45). Дружелюбное расположение и частые приступы смеха напоминают синдром Ангельмана. Возраст описанных пациентов достигает 3-26 лет. Кандидатным геном заболевания рассматривается ген микротубулин-ассоциированного τ-протеина (MAPT, 17q21.31), миссенсмутации и мутации сплайсинга в котором идентифицированы при аутосомнодоминантной лобно-височной деменции с паркинсонизмом.
Спектр других хромосомных регионов, микроделеции которых приводят к развитию известных в настоящее время микроделеционных синдромов, включает сегменты 8q21.3-q22.1, 16p11-p12.1, 17q11.2-q12, 17q12, 17q21.31 и, скорее всего, будет пополнен в ближайшем будущем. Что касается микродупликационных синдромов, то их регистрируют значительно реже. Как правило, они характеризуются более мягкими клиническими признаками. Так, можно отметить синдром частичной трисомии 3q29, возникающий вследствие микродупликации размером 1,61 млн пар нуклеотидов, затрагивающей гены PAK2 и DLG1. Заболевание характеризуется легкой или умеренной степенью умственной отсталости, ожирением и микроцефалией. Пациенты имеют круглое лицо, высокие изогнутые брови, широкую переносицу, бульбовидный нос, крупные глаза, уши и рот.
Можно отметить, что клиническая картина большинства отмеченных «новых» микроделеционных и микродупликационных синдромов, связанных с небольшими по размеру аберрациями хромосом, имеет, тем не менее, классические черты хромосомных заболеваний, включая умственную отсталость различной степени тяжести, комплекс дисморфических изменений лица, конечностей и пороков развития внутренних органов. В связи с этим идентификация таких тонких хромосомных нарушений позволяет обнаруживать новые кандидатные гены, изменение числа копий которых сопряжено с формированием тех или иных клинических признаков. В конечном итоге это позволяет приблизиться к пониманию ключевых патогенетических механизмов возникновения хромосомных болезней. С практической точки зрения возможность развития клинической картины хромосомного заболевания вследствие микроструктурных перестроек хромосом диктует строгую необходимость применения молекулярно-цитогенетических методов для их диагностики.
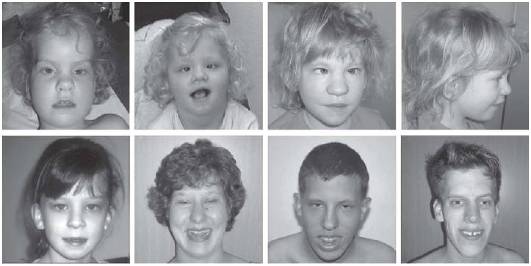
Рис. 22-45. Характерные лицевые аномалии у пациентов с микроделецией 17q21.31 (Slavotinek A.M. Novel microdeletion syndromes detected by chromosome microarrays // Hum. Genet. - 2008. - Vol. 124. - P. 1-17). Автор использовал иллюстрации из трех работ с разрешения издательств Macmillan Publishers Ltd и Nature Publishing Group: Koolen D.A., Vissers L.E., Pfundt R. et al. A new chromosome 17q21.31 microdeletion syndrome associated with common inversion polymorphism // Nat. Genet. - 2006. - Vol. 38. - P. 999-1001; Shaw-Smith C., Pittman A.M., Willatt L. et al. Microdeletion encompassing MART at chromosome 17q21.3 is associated with developmental delay and learning disability // Nat. Genet. - 2006. - Vol. 38. - P. 1032-1037; Sharp A.J., Hansen S., Selzer R.R. et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome // Nat. Genet. - 2006. - Vol. 38. - P. 1038-1042; верхний ряд, слева направо фотография из: 1 - Koolen et al., 2006; 2 - Shaw-Smith et al., 2006; 3, 4 - Sharp et al., 2006; нижний ряд, слева направо фотографияиз: 1 - Shaw-Smith et al., 2006; 2 - Koolen et al., 2006; 3 - Shaw-Smith et al., 2006; 4 - Koolen et al., 2006.
СПИСОК РЕКОМЕНДУЕМОЙ ЛИТЕРАТУРЫ
Баранов В.С., Кузнецова Т.В. Цитогенетика эмбрионального развития человека: Научнопрактические аспекты. - СПб., 2007. - 640 с.
Бочков Н.П., Пузырев В.П., Смирнихина С.А. Клиническая генетика: Учебник. - М.: ГЭОТАР-Медиа, 2011. - 544 с.
Ворсанова С.Г., Юров Ю.Б., Чернышов В.Н. Медицинская цитогенетика: Учебное пособие. - М.: Медпрактика-М, 2006. - 300 с.
Гинтер Е.К. Медицинская генетика: Учебник. - М.: Медицина, 2003. - 448 с.
Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. - М.: Т-во научных изданий КМК; Авторская академия, 2007. - 448 с.
Назаренко С.А. Изменчивость хромосом и развитие человека. - Томск: Изд-во Том. ун-та, 1993. - 200 с.
Назаренко С.А., Яковлева Ю.С. Цитогенетика человека и хромосомные болезни: Методическое пособие. - Томск: STT, 2001. - 83 с.
Daniley M., Barkai G., Goldman B. et al. Detection of numerical chromosomal aberration by comparative genomic hybridization // Prenat. Diagn. - 1999. - Vol. 19. - P. 100-104.
Dean J., Cohen G., Kemp J. et al. Karyotype 69,XXX/47,XX,+15 in a 2,5-year-old child // J. Med. Genet. - 1997. - Vol. 34. - P. 246-249.
De Vries B.B.A., Winter R., Schinzel A., van Ravenswaaij-Arts. Telomeres: a diagnosis at the end of the chromosomes // J. Med. Genet. - 2003. - Vol. 40. - P. 385-398.
Gardner R.J.M., Sutherland G.R. Chromosome abnormalities and genetic counseling. - New York: Oxford University Press, 2004. - 577 p.
Garzicic B., Guc-Scekic M., Pilic-Radivojevic G. et al. A case of monosomy 21 // Ann. Genet. - 1988. - Vol. 31. - P. 247-249.
Griffin D.K. The incidence, origin and etiology of aneuploidy // Int. Rev. Cytol. - 1996. - Vol. 167. - P. 263-296.
Hassold T., Hunt P. To err (meiotically) is human: the genesis of human aneuploidy // Nat. Rev. Genet. - 2001. - Vol. 2. - P. 280-291.
Lebedev I.N., Ostroverkhova N.V., Nikitina T.V. et al. Features of chromosomal abnormalities in spontaneous abortion cell culture failures detected by interphase FISH analysis // Eur. J. Hum. Genet. - 2004. - Vol. 12. - P. 513-520.
Prandini P., Deutsch S., Lyle R. et al. Natural gene-expression variation in Down syndrome modulates the outcome of gene-dosage imbalance // Am. J. Hum. Genet. - 2007. - Vol. 81. - P. 252-263.
Reddy K.S. Double trisomy in spontaneous abortions // Hum. Genet. - 1997. - Vol. 101. - P. 339-345.
Reddy K.S. Triple aneuploidy in spontaneous abortions // Clin. Genet. - 1999. - Vol. 56. - P. 103-104.
Robinson W.P., Bernasconi F., Lau A., McFadden D.E. Frequency of meiotic trisomy depends on involved chromosome and mode of ascertainment // Am. J. Med. Genet. - 1999. - Vol. 88. - P. 34-42.
Schinzel A. Catalogue of Unbalanced Chromosome Aberrations in Man. - Second revised and expanded edition. - Berlin; New York: Walter de Gruyter, 2001. - 966 p.
Slavotinek A.M. Novel microdeletion syndromes detected by chromosome microarrays // Hum. Genet. - 2008. - Vol. 124. - P. 1-17.
Stankiewicz P., Lupski J.R. Genome architecture, rearrangements and genomic disorders // Trends Genet. - 2002. - Vol. 18. - P. 74-82.
Stefanova I., Jenderny J., Kaminsky E. et al. Mosaic and complete tetraploidy in live-born infants: two new patients and review of the literature // Clin. Dysmorphol. - 2010. - Vol. 19. - P. 123-127.
Yu C.W., Bock H.G., North E. et al. Aneuploidy of 8 autosome and polyploidy mosaicism in a live-born infant // Am. J. Hum. Genet. - 1995. - Vol. 57. - 131 p.