Наследственные болезни : национальное руководство / Под ред. Н. П. Бочкова, Е. К. Гинтера, В. П. Пузырева - Москва : ГЭОТАР-Медиа, 2012. - 936 с. (Серия "Национальные руководства") - ISBN 978-5-9704-2231-1 |
Аннотация
Национальные руководства - первая в России серия практических руководств по основным медицинским специальностям, включающих всю основную информацию, необходимую врачу для непрерывного последипломного образования.
Национальное руководство "Наследственные болезни" содержит актуальную, современную информацию о геноме человека, общих вопросах медицинской генетики, клинической генетике. Руководство состоит из двух частей, в которых излагаются теоретические и клинические вопросы медицинской генетики. В первой части представлены новейшие данные по теоретическим вопросам медицинской генетики. Сведения об организации и функциях генома, генов и хромосом изложены в понятной для врачей форме, но без излишнего упрощения. Во второй части представлены вопросы клинической генетики, а именно методы диагностики наследственных болезней (от клинического уровня до секвенирования ДНК и РНК), принципов лечения и профилактики отдельных нозологических форм. Поскольку в национальных руководствах по другим специальностям описаны многочисленные наследственные болезни, на них можно найти ссылки [см. "Перечень наследственных болезней (синдромов), описание которых представлено в других национальных руководствах" на компакт-диске]. Приложение к руководству на компакт-диске включает более полную информацию по некоторым главам, электронную версию руководства, обширный иллюстративный материал, приложения, перечень наследственных болезней, описанных в других руководствах, фармакологический справочник. В подготовке настоящего издания в качестве авторов-составителей и рецензентов принимали участие ведущие ученые разных специальностей: генетики, иммунологи, невропатологи, фармакологи, онкологи и другие специалисты. Все рекомендации прошли этап независимого рецензирования.
Руководство предназначено для врачей-генетиков, врачей лаборантов-генетиков, врачей смежных специальностей, интернов, ординаторов, аспирантов, особенно по таким дисциплинам, как педиатрия, акушерство-гинекология, нервные болезни.
Гриф
Национальное руководство рекомендовано Российским обществом медицинских генетиков и Ассоциацией медицинских обществ по качеству
Глава 29. Медико-генетическое консультирование
Институт медико-генетических консультаций начал формироваться во всех странах, в первую очередь в США и Великобритании, после Второй мировой войны, хотя его истоки относятся к гораздо более раннему времени. Впервые сведения о типах наследования некоторых болезней и вероятности их повторения в семье встречаются в книге лондонского врача Д. Адамса «Философский трактат о наследственных свойствах человеческой расы» (1814). Принципы медико-генетического консультирования в современном понимании были изложены в серии публикаций американского генетика Шелдона Рида (1947, 1955), в которых он сформулировал цели и задачи, содержание медико-генетического консультирования и определил его как социальную службу, направленную на помощь семье без относительного влияния на общество и политику.
Начало становления медико-генетического консультирования в нашей стране связано с деятельностью С.Н. Давиденкова, который еще в конце 20-х гг. XX в. впервые в мире не только теоретически сформулировал, но и реализовал на практике принципы организации медико-генетических консультаций как центров профилактики наследственных болезней. Таким образом, медико-генетическое консультирование следует рассматривать как один из видов специализированной помощи населению, направленный главным образом на предупреждение появления в семье больных с наследственной патологией.
Рабочий комитет Американского общества генетики человека (1975) определил медико-генетическое консультирование как «?коммуникативный процесс, связанный с решением проблем, относящихся к появлению или риску появления наследственных болезней в семье. Этот процесс заключается в попытке одного или нескольких квалифицированных специалистов объяснить пациенту или семье диагноз, тип наследования, основные проявления, течение и доступное лечение наследственного заболевания; помочь семье принять определенное решение относительно репродуктивного поведения с учетом величины повторного риска и выбрать ряд действий в соответствии с этим решением, учитывая степень риска и семейные цели; помочь обратившимся лучше адаптироваться к наличию больного в семье и риску повторения этой болезни». В последующие годы определение генетического консультирования уточнялось, и последнее определение было предложено в 2006 г. специальной группой Национального общества генетиков-консультантов и одобрено Советом директоров Общества: «?генетическое консультирование - процесс помощи людям в понимании и адаптации к медицинским, психологическим и семейным особенностям генетического вклада в болезнь. Этот процесс включает следующие элементы: интерпретацию семейной и медицинской истории для оценки шансов появления или повторения заболевания; обучение относительно принципов наследования, тестирования, помощи, профилактики; информацию о ресурсах и исследованиях; консультирование с целью помочь сделать информированный выбор и адаптироваться к риску и состоянию болезни».
Из этих определений формируются основные задачи медико-генетического консультирования:
Таким образом, перед медико-генетическими консультациями стоят две главные цели, одна из которых заключается в предотвращении появления больного в семье, вторая - не менее важная - в помощи обратившимся пациентам понять информацию о диагнозе, прогнозе и адаптироваться к ситуации.
ПОКАЗАНИЯ ДЛЯ НАПРАВЛЕНИЯ В МЕДИКО-ГЕНЕТИЧЕСКУЮ КОНСУЛЬТАЦИЮ
Большинство пациентов и их семьи (примерно 85-95%) направляются в медико-генетические консультации врачами разных специальностей для уточнения диагноза и прогноза потомства. Даже если диагноз наследственного заболевания при постановке не вызывал у специалиста никаких сомнений, то пациент и его семья в обязательном порядке должны быть направлены к врачу-генетику для оказания дальнейшей помощи семье: определения величины риска появления такого же заболевания для следующего ребенка в этой семье, а также для других родственников. Например, если невролог поставил ребенку диагноз «мышечная дистрофия Дюшенна», то врач-генетик на основе этого диагноза (иногда с помощью генетических методов исследования приходится уточнять диагноз) может определить величину риска появления такого же заболевания для следующего ребенка в этой семье, а также для других членов семьи (сестры матери, сестры пробанда). Более того, в медико-генетической консультации можно осуществить пренатальную диагностику с помощью разных методических подходов и лабораторных методов, чтобы предотвратить рождение больного ребенка. Таким образом, объем оказываемой помощи расширяется. Если же врач не может точно поставить диагноз, но по некоторым признакам предполагает наличие наследственного заболевания, он должен направить больного и его семью в медикогенетическую консультацию с целью уточнения диагноза и прогноза для жизни больного и его потомства.
Ниже приводятся основные признаки, по которым можно заподозрить у пациента наследственную патологию. Эти признаки базируются на некоторых общих закономерностях, характеризующих проявление генов вообще и мутантных генов в частности.
Вовлеченность в патологический процесс нескольких органов (полисистемность поражения) . Такая особенность клинической картины большинства (если не всех) наследственных болезней связана с независимым, автономным проявлением действия гена в различных тканях, т.е. плейотропным, или множественным, действием гена.
Сегрегация симптомов заболевания в семье по определенным правилам. Обычно менделевскую сегрегацию обнаруживает комплекс симптомов (ядро признаков), входящих в синдром. В этом случае врач видит в семье несколько больных с аналогичной клиникой.
Наличие у больного нескольких микропризнаков или нормальных вариантов фенотипа, имеющих диагностическое значение , т.е. входящих в минимальные диагностические признаки синдрома. Они могут обнаруживаться в различных органах, формирующихся из разных зародышевых листков. Диагностическая значимость микропризнаков увеличивается, если они выявляются в определенном сочетании друг с другом. Микропризнаки рассматриваются как маркеры наследственных и тератогенных синдромов.
Недоразвитие или чрезмерное развитие отдельных частей тела. Это связано с нарушением взаимодействия между зачатками органов или клеточными слоями в онтогенезе и другими процессами, следствием чего является развитие более или менее специфических симптомов наследственной патологии, зависимой от времени проявления мутантного гена.
Согласованность времени манифестации и характера нарушения с этапами онтогенеза. Разброс во времени начала проявлений при наследственных заболеваниях значительно уже, чем при ненаследственных. Появление ряда симптомов наследственных болезней строго «привязано» к возрасту больного, и, таким образом, время начала заболевания выступает в качестве дополнительного диагностического признака.
Прогрессирование заболевания, отсутствие эффекта от лечения. Для наследственной патологии нетипично улучшение состояния при применении адекватной терапии. Редкими исключениями являются патогенетическая терапия и генотерапия.
Объективное обследование пробанда и его родственников, включающее изучение фенотипа, антропометрию, результаты специальных генетических методов, позволяет врачу-генетику определить точный диагноз. Для определения прогноза потомства в семье врач-генетик использует разные методы расчета генетического риска.
Самой частой причиной обращения в медико-генетическую консультацию является рождение у здоровых родителей ребенка с наследственной болезнью или врожденным пороком развития. Родители хотят знать прогноз здоровья для следующего ребенка в этом или другом браке - это так называемое ретроспективное консультирование. На втором месте стоят обращения для уточнения диагноза при подозрении на наследственную патологию у ребенка или взрослого с целью выбора адекватного способа лечения или реабилитации. Третья группа консультирующихся состоит из здоровых людей, имеющих родственников с наследственным заболеванием и желающих знать прогноз здоровья для себя и своих детей, - это так называемое проспективное консультирование. В последние годы в медикогенетических консультациях довольно существенный поток составляют семьи с новорожденными, у которых при неонатальном скрининге выявляются некоторые наследственные болезни обмена веществ (в настоящее время в нашей стране проводится массовое обследование всех новорожденных на наличие пяти заболеваний: фенилкетонурию, врожденный гипотиреоз, адреногенитальный синдром, муковисцидоз и галактоземию), и беременные, у которых в результате пренатального скрининга выявлена та или иная патология плода.
ЗАДАЧА МЕДИКО-ГЕНЕТИЧЕСКИХ КОНСУЛЬТАЦИЙ С ТОЧКИ ЗРЕНИЯ ОРГАНИЗАЦИИ ЗДРАВООХРАНЕНИЯ
Теоретические расчеты показывают, что суммарная величина генетического груза достигает 0,2 от условной единицы; это означает, что 1/5 всего генофонда современных популяций подвержена таким мутациям, которые проявляются как пре- и постнатальная смертность, врожденные пороки развития, наследственные болезни, бесплодие . Величина генетического груза значительно колеблется от популяции к популяции, что обусловлено действием факторов популяционной динамики, таких как инбридинг, миграция, дрейф генов, новые мутации и селекция, нарушающих величины генных частот. Наиболее значимы для медикогенетического консультирования первые три фактора.
Инбридинг, отклонение от случайного вступления в брак в сторону кровнородственных браков, - главный фактор, нарушающий равновесие генотипов в популяции. Инбридинг является частным случаем ассортативных браков. Браки между родственниками встречаются в разных популяциях с разной частотой, что приводит к разной отягощенности наследственными болезнями, и в первую очередь с аутосомно-рецессивным типом наследования. Генетическое значение инбридинга состоит в постепенном увеличении доли гомозиготных людей и уменьшении доли гетерозиготных людей в популяции, т.е. в инбредной популяции редкие рецессивные гены оказываются в гомозиготном состоянии чаще, чем в панмиксной популяции. Причем чем реже ген в популяции, тем больше роль инбридинга в манифестации заболевания.
Миграция выступает как фактор, по эффекту противоположный инбридингу. Миграционный поток приводит к нарушению границ браков, способствует выравниванию генных частот и гетерозиготизации населения. Однако миграция может привести и к накоплению генов в гомозиготном состоянии, когда мигрируют семьи, группы родственников, кланы.
Генетический дрейф действует аналогично инбридингу. В малых популяциях имеется большая вероятность случайного распределения генов. Поскольку генотипы потомства определяются генотипами родителей, то чем меньше популяция, тем меньше выбор для родительских гамет, тем интенсивнее протекает в ней процесс дрейфа, что приводит к снижению частоты одних аллелей и увеличению и закреплению других (например, финские, еврейские болезни и т.п.).
Первоочередная задача, стоящая перед медико-генетическими консультациями и органами здравоохранения, заключается в оценке величины генетического груза в регионе, т.е. определении доли людей, нуждающихся в помощи врача-генетика, а в широком смысле - в создании легко доступной сети генетической помощи для всех, кто в ней нуждается. Для этого целесообразен региональный подход к организации медико-генетической службы, основанный на изучении структуры популяции, величины груза наследственной патологии и его качественной характеристики. При планировании объема медико-генетической помощи и определении связи между числом консультаций и размером популяции в регионе следует учитывать вышеперечисленные факторы, нарушающие равновесие генных частот, от которых зависит в конечном счете отягощенность населения наследственной патологией. В этом случае медико-генетическая помощь будет действительно, с одной стороны, легко доступной, а с другой - обеспечит полный объем работы врача-генетика.
ЗАДАЧА МЕДИКО-ГЕНЕТИЧЕСКИХ КОНСУЛЬТАЦИЙ С МЕДИЦИНСКОЙ ТОЧКИ ЗРЕНИЯ
Основной задачей врача-генетика, работающего в консультации, является составление медико-генетического прогноза в семье и выбор профилактических мероприятий для предотвращения рождения больного ребенка. В понятие «медико-генетический прогноз» включаются по крайней мере три элемента: определение степени генетического риска; оценка тяжести медицинских и социальных последствий той аномалии, по поводу которой консультируется пациент или семья; перспектива применения методов пренатальной диагностики и профилактического лечения. Правильное составление медико-генетического прогноза зависит от следующих факторов: точности диагноза, знания новейших литературных данных по специальности, адекватности применения методов расчета генетического риска.
Схема медико-генетического консультирования представлена на рис. 29-1.
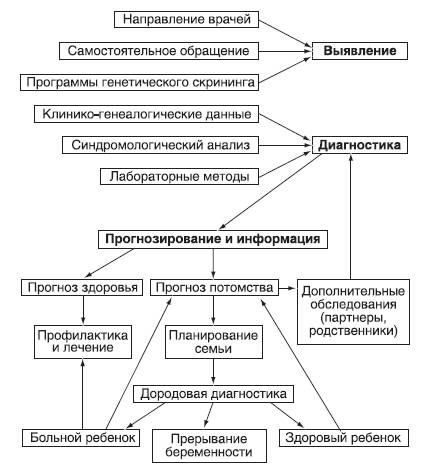
Рис. 29-1. Схема медико-генетического консультирования
Основной поток обращающихся в медико-генетические консультации формируется по направлениям врачей разных специальностей, в первую очередь педиатров, так как примерно 70-80% наследственной и врожденной патологии проявляется в детском возрасте, а также по результатам скрининга на некоторые наследственные болезни и врожденные пороки развития.
Точный диагноз заболевания у пробанда является абсолютно необходимым условием для любой консультации, так как на его основе базируется генетический прогноз для всей семьи. Следует учитывать, что при наследственных заболеваниях, в силу некоторых генетических закономерностей, в патологический процесс могут вовлекаться практически все системы и органы человека, так как мутантные гены автономно экспрессируются в любой ткани. Как уже говорилось, один ген часто проявляет множественные эффекты (плейотропное действие гена), в результате чего поражаются одновременно несколько органов. Именно поэтому врач-генетик использует, как правило, синдромологический подход к диагностике заболевания, пытаясь объединить все имеющиеся у больного симптомы на основе единого генеза. При этом он нередко прибегает к консультациям врачей самых разных специальностей: окулистов, дерматологов, невропатологов и др. Кроме того, медико-генетические консультации располагают большим арсеналом специфических методов, позволяющих уточнить диагноз, главные из которых - клинико-генеалогический, цитогенетический, специальные биохимические методы, а в последнее время и методы ДНК-диагностики. Достижения последних лет в генетике, особенно после завершения секвенирования генома человека (2003 г.), внесли заметный вклад в понимание этиологии и патогенеза заболеваний человека, в обнаружение тех или иных молекулярных механизмов, приводящих к появлению клинических симптомов заболевания. Поскольку в настоящее время гены большинства наследственных болезней картированы, выделены и клонированы, диагностические возможности, включая пресимптоматическую диагностику, значительно расширяются, а следовательно, увеличивается точность прогнозирования.
Генетический риск - это вероятность появления определенной наследственной патологии у обратившегося за консультацией или у его потомков. Он определяется путем расчетов, основанных на генетических закономерностях, или с помощью эмпирических данных. Возможность рассчитать генетический риск зависит в основном от точности диагноза и полноты генеалогических данных. Большое значение имеет выявление кровного родства между консультирующимися супругами; изучение состояния здоровья монозиготных близнецов, если такие есть в родословной; обследование как больных, так и здоровых членов семьи; сведения о выкидышах, мертворождениях. Правильный сбор и анализ родословной иногда дают возможность провести консультацию даже в тех случаях, когда диагноз точно установить не удается.
Генетический риск до 5% оценивается как низкий и не считается противопоказанием к деторождению в данной семье. Риск от 6 до 20% считается средним; в этом случае решение семьи относительно планирования дальнейших беременностей зависит не только от величины риска, но и от тяжести медицинских и социальных последствий конкретного наследственного заболевания, а также от возможности пренатальной диагностики. Генетический риск, превышающий 20%, относится к категории высокого, и при отсутствии методов пренатальной диагностики соответствующей патологии решение о дальнейшем продолжении или прекращении деторождения в семье бывает трудным.
Клинико-генеалогический метод является основным аналитическим методом в практике медико-генетического консультирования. Он применяется в следующих целях: для установления наследственного характера заболевания, для определения типа наследования и пенетрантности патологического гена, для анализа генетического сцепления, при расшифровке механизмов взаимодействия генов.
Первые попытки сбора генеалогических сведений содержатся в Библии: «родословие» Иисуса Христа (Авраам родил Исаака; Исаак родил Иакова и т.д.). Известный историк В.М. Соловьев составил обширные родословные русских царей, используя имена. Ф. Гальтон в книге «Наследование таланта» при составлении родословных известных людей использовал фамилии. Система условных графических обозначений в родословных впервые появилась в 1931 г. (Г. Юст). Условно унифицированная система принята в 1991 г. Комитетом по генетическому консультированию Американского общества генетики человека. В последующие годы в нее вносятся некоторые дополнения.
Технически клинико-генеалогический метод складывается из двух этапов: составления родословной (сбор сведений и графическое их изображение) и ее анализа.
Составление родословной Начинать сбор родословной следует от пробанда, т.е. больного (живого или умершего), из-за которого возникла необходимость обращения за медикогенетической консультацией. В генетическую карту подробно записываются сведения о больном. В дальнейшем продолжают сбор данных о сибсах (братьях и сестрах) пробанда, его родителях, о сибсах родителей, их детях и т.д. Врач должен собрать не только сведения, касающиеся конкретного патологического признака в семье, но и информацию о других серьезных заболеваниях и аномалиях, встречающихся среди членов семьи. Важно получить сведения о спонтанных абортах, мертворожденных и ранней детской смертности. Иногда они могут иметь прямое отношение к проблеме и сыграть важную роль при оценке прогноза. Всегда нужно фиксировать хотя бы основные данные с обеих сторон (по отцовской и материнской линии), даже если речь идет об аутосомно-доминантном заболевании, унаследованном от одного из родителей.
На основании собранных сведений строят графическую родословную. Наиболее распространенные символы, используемые в родословных, представлены на рис. 29-2.
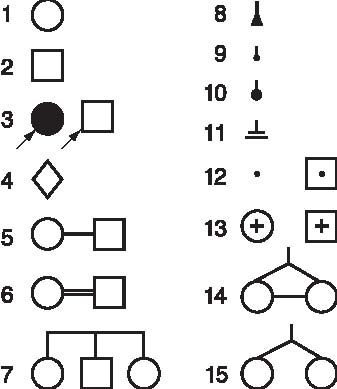
Рис. 29-2. Символы, используемые при составлении родословных: 1 - женский пол; 2 - мужской пол; 3 - пробанд; 4 - пол неизвестен; 5 - брак; 6 - родственный брак; 7 - сибсы; 8 - спонтанный аборт; 9 - медицинский аборт; 10 - мертворожденный; 11 - бездетный брак; 12 - гетерозиготные носители мутантного гена; 13 - умершие; 14 - монозиготные близнецы; 15 - дизиготные близнецы
Все полученные данные о состоянии здоровья родственников, причинах и возрасте смерти записываются под родословной (легенда). Желательно получить надежные и достоверные сведения о родственниках (по возможности, в 3-4-м поколении). Необходимо указывать дату составления родословной. Целесообразно указывать девичьи фамилии женщин: это особенно важно для X-сцепленных заболеваний, поскольку паспортная фамилия членов родословной может меняться в каждом поколении. Полезно записывать адреса некоторых членов семьи - это может помочь в сборе дополнительной информации, а также при последующих контактах с родственниками. Поколения принято обозначать римскими цифрами сверху вниз (слева от родословной). Арабскими цифрами обозначается потомство одного поколения (весь ряд слева направо последовательно). Желательно, чтобы братья и сестры располагались в родословной по порядку рождения. Таким образом, каждый член родословной имеет свой уникальный номер, например I3, II5, III8 и т.д. Важно отметить в родословной лично обследованных людей на наличие заболевания (символ!). Врач должен стремиться к получению наиболее объективного первичного материала, который закладывается в основу генетического анализа родословной. Сбор родословной должен проходить в спокойной непринужденной обстановке, без посторонних людей. Можно собирать родословную по анкете, но лучше лицом к лицу, лично. Иногда данные способы комбинируются.
Анализ родословной Целью анализа родословной является установление генетических закономерностей передачи заболевания. Правильно собранная родословная помогает ответить на следующие вопросы.
Встречалось ли аналогичное заболевание у родственников больного?
Обнаруживается ли в родословной другое заболевание, имеющее диагностическое или прогностическое значение?
Достаточно ли имеется данных для установления типа наследования?
Какие дополнительные мероприятия нужны для завершения полноценного клинико-генеалогического анализа (получение медицинских документов с точными клиническими сведениями о больных, проведение дополнительных клинических и лабораторных исследований и т.д.)?
В рамках клинико-генеалогического анализа важно учитывать такие характеристики наследственных заболеваний, как пенетрантность и экспрессивность мутантного гена.
Пенетрантность и экспрессивность Одни гены почти не проявляют изменчивости в своем фенотипическом выражении, для проявления других генов характерна высокая степень изменчивости. Такая изменчивость, с одной стороны, может быть обусловлена тем, что не все люди, имеющие данный генотип, имеют соответствующий ему фенотип, а с другой - тем, что степень проявления фенотипа может быть различной у разных людей. Пенетрантность гена - это доля людей с определенным генотипом, у которых ожидаемый фенотип проявляется на клиническом уровне, от общего числа людей с данным генотипом. Экспрессивность гена - степень выраженности фенотипа у тех людей, у которых он вообще проявился.
Понятие неполной пенетрантности на практике чаще всего применимо к аутосомно-доминантным заболеваниям. Особенность таких заболеваний заключается в том, что иногда у заведомых носителей гена, имеющих пораженных родителей и детей, признаки заболевания отсутствуют (бессимптомные гетерозиготы). В таких случаях речь идет о так называемых пропусках (проскоках) поколений, что и является прямым доказательством неполной пенетрантности. Можно сказать, что пенетрантность - это частота фенотипического проявления гена.
Самый простой метод определения пенетрантности заключается в определении фактической доли больных потомков в тех браках, где один из супругов болен, а другой здоров. По генотипической характеристике такие браки относятся к типу Aa × aa, и в них ожидаемая доля больных детей составляет 50%. Если фактическое число больных детей оказывается меньше ожидаемых 50%, можно предположить наличие неполной пенетрантности. Численная оценка коэффициента пенетрантности может быть выражена как отношение наблюдаемого числа больных детей в браках типа Aa × aa к ожидаемому числу больных при 50% вероятности унаследования патологического генотипа. Однако следует отметить, что расчеты такого типа применимы не к отдельным семьям небольшого размера, где соотношение больных и здоровых подвержено значительной случайной вариации, а только к большим выборкам семей с однотипной патологией.
Более надежным считается так называемый прямой способ оценки пенетрантности по пропускам поколений. При таком подходе в родословной (или в выборке однотипных родословных) необходимо выбрать все звенья, содержащие больных в поколении дедов и поколении внуков, т.е. трехчленные цепочки прямой передачи гена. После подсчета общего числа таких цепочек проводится подсчет числа людей с заболеванием и без симптомов заболевания в промежуточном (втором) поколении. Соотношение числа больных к общему числу людей в этом поколении (т.е. к числу цепочек) и соответствует коэффициенту пенетрантности. Классическим примером заболеваний с полной пенетрантностью является ахондроплазия. Из распространенных заболеваний с неполной пенетрантностью можно упомянуть такие как нейрофиброматоз, синдром Марфана.
Варьирующая экспрессивность мутантных генов ярко проявляется при многих наследственных заболеваниях с любым типом наследования. Так, при классическом доминантном варианте несовершенного остеогенеза в одной и той же семье могут наблюдаться как полная клиническая картина заболевания (ломкость костей, глухота, голубая окраска склер), так и минимальные проявления (например, только голубые склеры). Это обстоятельство важно учитывать, поскольку даже у обладателя минимальных фенотипических проявлений болезни риск передачи этого заболевания потомству, в том числе и в полной клинической форме, составляет 50%. При анализе родословной любой ее член, связанный родством с больными и имеющий хотя бы минимальные симптомы, считается гетерозиготой по доминантному гену заболевания.
Основной задачей врача-консультанта при медико-генетическом консультировании является определение прогноза потомства в обратившейся семье. Расчет повторного генетического риска базируется на теории вероятностей. Согласно классическому определению вероятность - это математическая, числовая характеристика, определяющая степень возможности появления какого-либо события при тех или иных условиях, которые могут повторяться неограниченное число раз. Численное значение вероятности определяется как отношение числа случаев, «благоприятствующих» данному событию, к общему числу «равновозможных» случаев. При консультировании семей с наследственными заболеваниями врачгенетик, опираясь на законы менделевской генетики и математические правила теории вероятностей, рассчитывает количественные оценки риска, которые и являются основой генетического прогноза. Риск может выражаться в виде простых и десятичных дробей, процентов или шансов. В практике медико-генетического консультирования при объяснении прогноза семьям чаще всего используется процентное обозначение величин риска, но в процессе вычислений удобнее пользоваться простыми дробями.
Существуют два принципиальных подхода к оценке генетического риска: теоретические расчеты, основанные на законах формальной генетики, и эмпирические данные (нередко в качестве повторного риска используются эмпирические показатели частоты заболевания среди родственников определенных степеней родства). Методология вероятностного прогнозирования при менделирующих и неменделирующих заболеваниях существенно различается. Для менделирующих заболеваний достаточно четко разработаны теоретические основы оценки генетического риска, поэтому основная задача сводится к идентификации генотипа, лежащего в основе заболевания, и вероятностной оценке так называемой сегрегационной частоты, в зависимости от генотипов родителей будущего ребенка. При сложно наследующихся заболеваниях консультирование часто основывается на методе «черного ящика», т.е. на чистом эмпиризме, поскольку при многофакторных заболеваниях, в принципе, невозможно устанавливать специфические дискретные генотипы, ответственные за развитие болезни. В подобной ситуации формальный генетический анализ, направленный на точное вероятностное прогнозирование, связан с применением специальных генетических моделей и сложных математических методов.
Расчет риска при моногенных заболеваниях При расчетах риска для заболеваний с любым типом наследования обычно подразумевается анализ двух возможных ситуаций:
В первом случае, когда генотипы консультирующихся известны (по анализу родословной удается идентифицировать генотипы обоих родителей) и известен тип наследования заболевания, оценка риска проводится в соответствии с простым менделевским расщеплением. Причем в рамках генеалогического анализа важно учитывать такие характеристики наследственных заболеваний, как пенетрантность и экспрессивность. На рис. 29-3 представлены фрагменты родословных и принципы оценки риска.
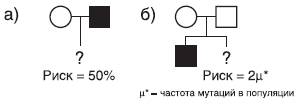
Рис. 29-3. Расчет генетического риска при известных генотипах родителей. Аутосомно-доминантное заболевание с полной пенетрантностью (например, ахондроплазия)
В первом фрагменте родословной (см. рис. 29-3а), основываясь на законах Г. Менделя, нетрудно определить, что генетический риск составляет 50%. Во втором фрагменте (см. рис. 29-3б) показано, что у двух здоровых супругов родился больной ребенок с ахондроплазией. Родители хотят узнать, какова вероятность того же заболевания для их следующего ребенка. Поскольку данное заболевание определяется геном со 100% пенетрантностью, то родители являются нормальными гомозиготами (тип брака aa × aa), а больной ребенок должен быть гетерозиготой (Aa). Происхождение патологического аллеля, приведшего к заболеванию, можно объяснить новой мутацией в половой клетке либо у отца, либо у матери (если не принимать в расчет возможность внебрачного зачатия). Частота спонтанных мутаций в популяции представляет собой малую величину (μ=10-4-10-6). Таким образом, вероятность рождения в этом браке еще одного больного ребенка составит 2μ (сумма вероятностей мутаций в сперматозоиде и яйцеклетке).
Большинство аутосомно-доминантных заболеваний проявляются с неполной пенетрантностью. Например, если при нейрофиброматозе пенетрантность равна 80%, то риск его повторения в данной семье равен не 50, а 40%. Пример такой родословной представлен на рис. 29-4.
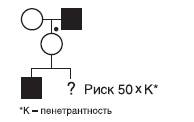
Рис. 29-4. Расчет генетического риска при известных генотипах родителей. Аутосомно-доминантное заболевание с неполной пенетрантностью
В традиционном понимании расчеты риска при аутосомно-рецессивных заболеваниях при известных генотипах родителей. принято считать относительно простыми. Если Аутосомно-доминантное заболевание у здоровых родителей имеется больной ребенок, то закономерно сделать вывод о том, что они оба являются носителями гена заболевания, и риск для каждого будущего ребенка в этом браке составляет 25% (1/4), при условии совпадения биологического и паспортного отцовства. Вероятность того, что здоровый сибс больного является носителем, составляет 2/3. Величина риска для других родственников больных (не сибсов), как правило, оказывается очень низкой. Пример родословной с аутосомно-рецессивным заболеванием представлен на рис. 29-5.
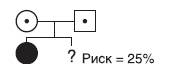
Рис. 29-5. Расчет генетического риска при известных генотипах родителей. Аутосомно-рецессивное заболевание
Женщины в этих двух родословных (рис. 29-6) являются облигатными носительницами, так как в первом случае женщина родила двух больных мальчиков, а во втором - больны дед и внук; вероятность новых мутаций в этих случаях ничтожно мала.
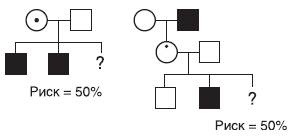
Рис. 29-6. Расчет генетического риска при известных генотипах родителей. X-сцепленное заболевание
При кровнородственных браках риск рождения больного ребенка, в первую очередь с аутосомно-рецессивным заболеванием, зависит от степени родственной близости супругов, мерой которой служит R - коэффициент родства (доля общих генов). Например, для двоюродных сибсов расчет риска проводится следующим образом: 1/8 (R) × 4 (латентная генетическая нагрузка в популяции) × 1/4 (аутосомно-рецессивное наследование) = 1/8 (12,5%). К полученной величине риска следует прибавить 5% общепопуляционного риска.
Для случаев, когда генотипы родителей известны, существует следующее правило: если априори на основании родословной удалось установить генотипы консультирующихся (тип брака), то никакая информация апостериори (с учетом имеющихся детей) не может изменить соответствующую вероятность величины риска. В той ситуации, когда генотипы консультирующихся неизвестны, возникают более сложные генетические задачи, решение которых невозможно без применения соответствующих математических расчетов, основанных на теории вероятностей. Чаще всего применяется теорема Байеса, представляющая собой метод сравнения всех вероятностей или возможных событий с их последующим взвешиванием за счет включения дополнительной информации, которая позволяет установить, какое из этих событий наиболее вероятно. В подобных случаях на сновании конкретной родословной выдвигается несколько рабочих гипотез о генотипах, и вероятность каждой из них оценивается с учетом не только априорной информации (по наследованию от родителей), но и апостериорной, связанной с рождением детей. На рис. 29-7 изображена родословная с Х-сцепленным заболеванием.
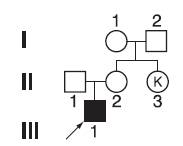
Рис. 29-7. Пример расчета генетического риска при неизвестных генотипах родителей
За консультацией обратилась тетя пробанда II3 в связи с мышечной дистрофией Дюшенна у племянника III1. Она хочет знать прогноз для своих детей. Поскольку речь идет об Х-сцепленном заболевании, то возможность носительства патологического гена для этой женщины зависит от того, является ли носительницей ее мать (I1). Можно предположить следующие генетические объяснения заболевания у III1:
I1 - гетерозиготная носительница аномального гена, передала этот ген дочери II2, которая, в свою очередь, передала его своему сыну III1;
I1 - гомозигота по нормальному аллелю, но она или ее муж передали новую мутацию дочери II2, которая, являясь гетерозиготой, передала ее сыну III1;
I1 и II2 - гомозиготы по нормальному аллелю, но новая мутация возникла в половой клетке у II2.
Только первая гипотеза исходит из того, что I1 является гетерозиготной носительницей, и только в этом случае консультирующаяся может оказаться носительницей аномального гена. По правилам теории вероятности сумма вероятностей (Р) всех гипотез равна 1, т.е. Р(А) + Р(В) + Р(С) = 1. Логику дальнейших рассуждений и вычислений удобнее всего представить в форме табл. 29-1.
Таблица 29-1. Расчет генетического риска при различных вариантах генотипа I1
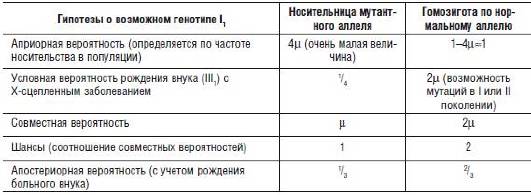
Итак, если для I1 вероятность носительства гена мышечной дистрофии Дюшенна составила 1/3, то для консультирующейся (ее дочери) она должна быть вдвое меньше, т.е. составлять 1/6 (примерно 16,5%). В конечном итоге риск рождения больного сына для II3 составляет 1/6 × 1/2 = 1/12 (т.е. приблизительно 8%).
При многих Х-сцепленных рецессивных заболеваниях, в том числе и при мышечной дистрофии Дюшенна, лабораторные тесты на носительство дают существенное перекрывание результатов для носителей и неносителей. Их использование значительно модифицирует риск. Так, при мышечной дистрофии Дюшенна уровень креатинфосфокиназы в сыворотке повышен примерно у 2/3 всех носительниц, а статистический анализ перекрывающихся кривых для носителей и неносителей позволяет получить условные вероятности носительства, основанные только на уровне креатинфосфокиназы. В нашем примере консультирующаяся II3 имела нормальный уровень креатинфосфокиназы. При включении этой информации в стандартную таблицу теоремы Байеса получаем вероятность носительства не 16,5, а 6%. Использование сцепленных маркеров при мышечной дистрофии Дюшенна для определения вероятного статуса носительства консультирующейся позволило уменьшить риск в десятки раз.
Этот конкретный пример иллюстрирует, насколько умело и внимательно надо совмещать общий анализ родословной с результатами биохимических тестов и анализа сцепленных маркеров для модификации риска носительства. Для II3 этот риск носительства априори был равен 50%. Анализ родословной позволил снизить оценку риска до 16,5%. Включение информации об уровне креатинфосфокиназы снизило риск в еще большей степени - до 6%. И наконец, исследование маркеров ДНК привело к разительному снижению этой величины менее чем до 1 шанса из 800.
Следует заметить, что в подобных ситуациях справедливо правило: последующее рождение детей у самой консультирующейся имеет большое значение для величины риска носительства - рождение больного сына однозначно установит ее генотип (носительство патологического аллеля), а появление одного или нескольких здоровых сыновей снизит вероятность того, что их мать является гетерозиготной носительницей. В таком случае вновь придется прибегать к расчетам с априорными, условными, совместными и апостериорными вероятностями по теореме Байеса.
Расчет риска при заболеваниях с варьирующим возрастом начала Некоторые аутосомно-доминантные заболевания проявляются не с момента рождения, а только в юношеском или даже во взрослом возрасте. Примеры подобных заболеваний приведены ниже. Генеалогические сведения о семьях, в которых имеются такие заболевания, за рубежом нередко содержатся в конфиденциальных и тщательно пополняемых генетических регистрах, что позволяет при необходимости учитывать и использовать эту важную информацию. Одно из наиболее полезных применений таких регистров заключается в том, что молодые члены семей, отягощенных наследственными заболеваниями такого рода, могут получить информацию о риске развития заболевания как для себя, так и для своих будущих детей.
Аутосомно-доминантные заболевания с варьирующим возрастом манифестации:
На рис. 29-8 обратившийся в консультацию юноша (II1) достиг 25-летнего возраста и хочет узнать вероятность того, что он унаследовал от своего умершего отца (I1) семейный аденоматозный полипоз, кодирующийся геном FAP.
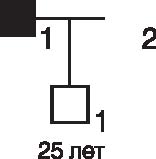
Рис. 29-8. Пример расчета риска при семейном аденоматозном полипозе
Очевидно, что II1 начал свою жизнь с априорной вероятностью унаследования гена заболевания 50% (1/2). Этот риск можно модифицировать по результатам обследования семейном аденоматозном полипозе кишечника, а также по наличию или отсутствию врожденной гипертрофии пигментного эпителия сетчатки (CHRPE). В данном случае оба обследования были проведены и дали отрицательные результаты (отсутствие признаков патологии).
Эта информация позволяет модифицировать априорную вероятность 50% (1/2) следующим образом. Вероятность того, что у 25-летнего гетерозиготного обладателя патологического гена при обследовании кишечника не будет обнаружено полипов, составляет 1 из 10. Вероятность того, что у него будет выявлено четыре или менее очага CHRPE, также равна 1 из 10 (Burn et al., 1991). Расчет по формуле Байеса проводится следующим образом:
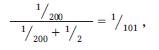
где 1/200 - совместная вероятность, 1/2 - априорная вероятность.
Таким образом, для II1 апостериорная вероятность унаследования гена FAP и развития заболевания в предстоящий период жизни составляет 1 шанс из 101 (менее 1%). Очевидно, что этот риск дает консультирующемуся более обнадеживающие перспективы, чем априорная вероятность 50%.
Расчет риска при заболеваниях с генетической гетерогенностью (разные типы наследования) При заболеваниях с доказанной генетической гетерогенностью, т.е. в таких случаях, когда идентичные по клиническим проявлениям наследственные болезни обусловлены мутациями в разных локусах и с разными типами наследования, расчет генетического риска представляет собой самостоятельную проблему. Здесь основные трудности связаны с оценкой риска в так называемых изолированных (единичных) случаях, в которых по родословной невозможно определить конкретный тип менделевского наследования. В подобных ситуациях расчет повторного риска (для детей или сибсов больного) необходимо проводить с учетом всех возможных типов наследования и региональных особенностей их распространения (популяционных частот). Расчет генетического риска в этом случае основан на вычислении полной вероятности, которая является суммой всех совместных вероятностей в байесовской таблице. Таким образом, оценки повторного риска для родственников первой степени в изолированных случаях заболевания определяются по частотному соотношению различных форм данного заболевания (априорная вероятность) и риску передачи заболевания при том или ином типе наследования (условная вероятность). Итоговая оценка риска (полная вероятность) рассчитывается как сумма всех совместных вероятностей.
Рассмотрим родословную с единичным случаем пигментной дистрофии сетчатки (рис. 29-9), взяв в качестве частотных характеристик разных типов наследования данные, полученные в ходе популяционных исследований в ивановской области.
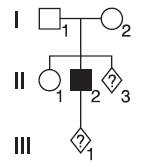
Рис. 29-9. Пример расчета риска при пигментной дистрофии сетчатки
Таблица 29-2. Частотное соотношение разных форм гетерогенных заболеваний и доля истинно спорадических случаев
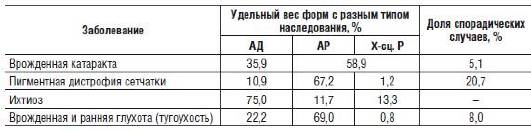
Примечание: АД - аутосомно-доминантный; АР - аутосомно-рецессивный; Х-сц. Р - рецессивный, сцепленный с X-хромосомой.
В данном примере риск заболевания для сибса больного (II3) составит:
(10,9×0)(67,2×0,25)(1,2×0,33)=17,2%.
В том же примере риск заболевания для ребенка больного (III1) составит:
(10,9×0,5)(67,2×0)(1,2×0)=5,45%.
Результаты расчетов для всех заболеваний, представленных в табл. 29-2, с учетом как пола пробанда, так и пола будущего родственника суммированы в табл. 29-3.
Таблица 29-3. Повторный риск для родственников больного в изолированных случаях заболеваний с генетической гетерогенностью
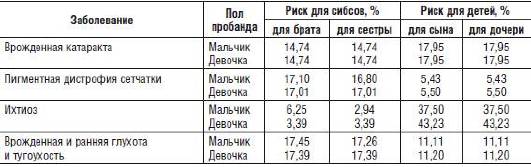
Таким образом, вышеизложенная информация - это попытка показать обобщенный подход к расчетам генетического риска при заболеваниях с различными типами менделевского наследования на основе законов классической генетики и основных правил теории вероятности. Важность этого раздела медикогенетического консультирования не вызывает сомнений, потому что бурный прогресс в молекулярно-генетических и других методах диагностики наследственных болезней, в том числе и дородовой, не должен настраивать врачей, занимающихся медико-генетическим консультированием, на пренебрежительное отношение к вероятностному прогнозу. Более того, именно на основе вероятностного прогнозирования определяются показания к дородовой диагностике, и расчеты вероятностей иногда являются ключевыми показателями для применения молекулярно-генетических методов.
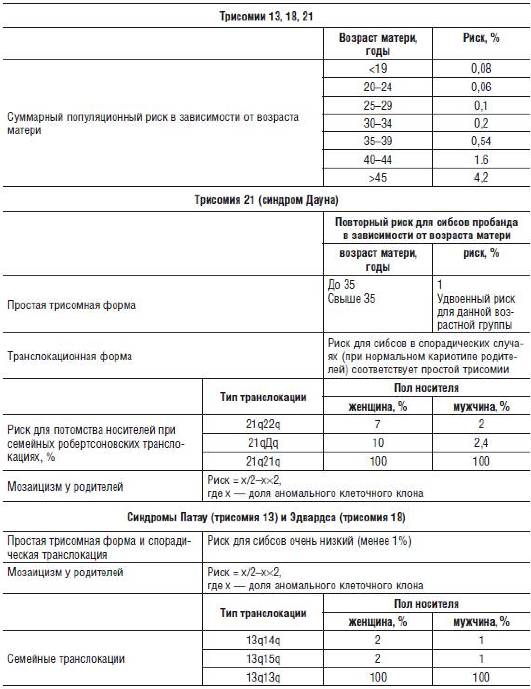
Расчет генетического риска при хромосомных болезнях Медико-генетическое консультирование при хромосомных аномалиях обычно не вызывает больших трудностей. Определение повторного риска проводится чаще всего в трех случаях:
В первом случае риск для сибсов пробанда оценивается на основании эмпирических данных для каждого типа аномалий и возраста матери. До 30 лет частота нерасхождений хромосом практически не увеличена; примерно 1% всех детей, рожденных матерями в возрасте 38-40 лет, имеют трисомию 21, и 3,7% - другие хромосомные аномалии.
При обнаружении мозаицизма у кого-либо из родителей пробанда риск для сибсов определяется по формуле
х/(2-х)×К,
где х - доля аномального клеточного клона; К - коэффициент элиминации несбалансированных зигот в эмбриогенезе. Например, при синдроме Дауна К=1/2.
Накопление в семье случаев хромосомной патологии можно объяснить структурными аномалиями хромосом родителей. В отличие от простой трисомии часто та транслокационных форм хромосомных синдромов не зависит от возраста матери и поэтому встречается относительно чаще у молодых родителей. Например, у 8% детей с синдромом Дауна, рожденных женщинами до 30 лет, отмечается транслокация; при этом в 2-3% случаев транслокация имеется также и у одного из родителей. У детей, рожденных женщинами более старшего возраста, транслокационный вариант болезни Дауна встречается лишь в 0,4% случаев. При семейных формах структурных аномалий хромосом можно теоретически определить процентное соотношение различных типов образующихся гамет и зигот. Однако наблюдаемая частота поражения детей ниже теоретически ожидаемой, что объясняется селекцией несбалансированных зигот в эмбриогенезе. Именно поэтому при семейных формах структурных аномалий хромосом риск тоже оценивается по эмпирическим данным. В усредненные эмпирические данные вносится расчетная поправка на величину перестроенного участка хромосомы. При наличии хромосомной перестройки у матери генетический риск, как правило, выше, чем при наличии такой перестройки у отца. Так, для распространенных транслокаций эмпирический риск составляет примерно 11%, когда носитель - мать, и 2%, когда носитель - отец. В крайне редких случаях транслокаций типа центрического слияния между двумя гомологичными хромосомами все гаметы имеют либо избыток, либо нехватку хромосомного материала. Именно поэтому теоретический и фактический риск для потомства носителя подобной транслокации равен 100%.
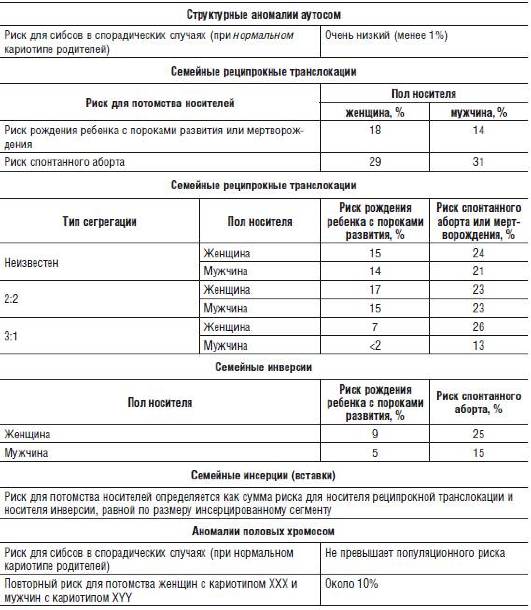
Сбалансированные структурные аномалии хромосом родителей могут быть причиной повторных спонтанных абортов. В этом случае риск невынашивания беременности зависит от пола носителя транслокации и характера перестройки.
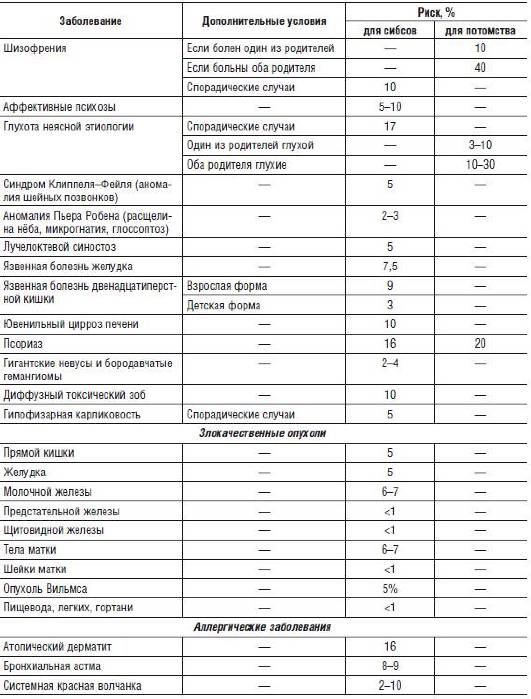
Если теоретическая оценка риска невозможна, применяют эмпирический подход. В практике консультирования используют таблицы эмпирического риска. Эмпирический риск для хромосомных болезней указан в табл. 29-4.
Таблица 29-4. Эмпирический риск при хромосомных болезнях
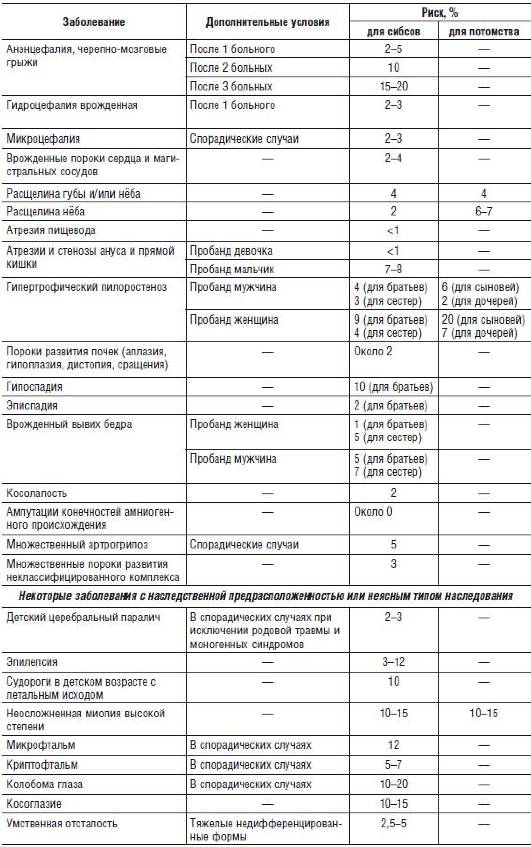
Расчет риска при мультифакториальных заболеваниях Болезни с наследственным предрасположением, или мультифакториальные заболевания, занимают ведущее место в наследственной патологии человека. Развитие большинства распространенных хронических болезней и врожденных пороков развития обусловлено совместным действием многих генов и факторов среды (сахарный диабет, язвенная болезнь, врожденные пороки сердца, дефекты невральной трубки). В таких случаях используют таблицы эмпирического риска, значение которого зависит от целого ряда факторов: семейной частоты, наследуемости признака, пола пробанда, тяжести поражения, формы заболевания (табл. 29-5).
Таким образом выглядит обобщенный подход к расчетам генетического риска при заболеваниях с различными типами менделевского наследования на основе законов классической генетики и основных правил теории вероятности, а также при хромосомных и мультифакториальных болезнях.
Таблица 29-5. Эмпирический риск при мультифакториальных заболеваниях
ЗАДАЧА МЕДИКО-ГЕНЕТИЧЕСКИХ КОНСУЛЬТАЦИЙ С СОЦИАЛЬНОЙ ТОЧКИ ЗРЕНИЯ
В настоящее время все большее внимание уделяется коммуникативной функции медико-генетического консультирования. Врач-генетик должен помочь консультирующимся понять медицинские факты, тип наследования заболевания, генетический риск его повторения в семье, лучше адаптироваться к несчастью и принять адекватное решение относительно дальнейшего деторождения. Эффективность медико-генетического консультирования напрямую зависит от грамотного выполнения этой коммуникативной функции врача-консультанта. Акценты взаимодействия врача-генетика и консультирующихся смещаются в сторону помощи семье в адаптации к генетическому риску или появлению наследственного заболевания у детей и родственников. В этих случаях основные усилия врача-генетика должны быть направлены на доходчивое, доступное построение заключительной беседы с ориентацией на разные образовательные уровни, структуру личности консультирующегося. Задача консультанта состоит не в том, чтобы обязательно добиться, например, прерывания беременности при обнаружении патологии плода. Консультант должен помочь семье правильно понять смысл медико-генетического заключения и принять адекватное решение относительно настоящей беременности и дальнейшего деторождения, не навязывая своего мнения.
Генетический риск следует интерпретировать таким образом, чтобы пациенты могли как можно лучше выбрать пути принятия решения, обеспечивающие им нормальный стиль жизни. Конечно, оптимальным вариантом для всех стран является такое рациональное решение консультирующихся, которое позволяет уменьшить бремя генетических болезней в семье и обществе, и большинство принимаемых решений бывает именно такими.
Но иногда принимаемые пациентами решения могут быть иррациональными с точки зрения здравого смысла. Например, семья, имеющая двух детей, больных муковисцидозом, принимает решение о рождении третьего больного ребенка, диагноз которому поставлен пренатально. В этом случае врач-генетик, базируясь на целях психологического благополучия консультирующихся, должен признать это решение как наиболее приемлемое для данной семьи, понять их поведение и помочь им в адаптации к этой ситуации. Каким бы ни было решение, принятое консультирующимися, врач-консультант должен полагаться на него как на наиболее подходящее в психологическом отношении для конкретной семьи. В любом случае необходимо стремиться найти пути к взаимному пониманию.
ЭФФЕКТИВНОСТЬ МЕДИКО-ГЕНЕТИЧЕСКОГО КОНСУЛЬТИРОВАНИЯ
Эффективность медико-генетического консультирования, если ее оценивать в терминах следования советам врача-генетика относительно дальнейшего деторождения, является, по данным большинства медико-генетических консультаций мира, довольно высокой и составляет 60-90%. Однако в настоящее время в работе врача-консультанта все большее внимание уделяется помощи семье в адаптации к генетическому риску или появлению наследственного заболевания у детей и родственников. Современное медико-генетическое консультирование направлено в первую очередь на достижение психолого-образовательной цели, т.е. на помощь семье в социальной адаптации. Именно поэтому определение медико-генетического консультирования в настоящее время может определяться как динамический психолого-образовательный процесс, который базируется на генетической информации. Отношения между врачом-генетиком и пациентами можно назвать терапевтическими, так как в их основе лежит помощь в понимании конкретной генетической информации, направленной на повышение способности консультирующихся адаптироваться, минимизировать психологический стресс. Помощь консультирующимся в таких психологических нарушениях, как чувство вины, снижение чувства собственного достоинства, социальная изоляция и др., является целью терапевтического консультирования. Таким образом, врач-консультант должен обладать многими качествами и быть:
-
квалифицированным врачом с опытом синдромологического подхода к диагностике;
-
генетиком, знающим формальные основы и современные достижения генетики;
-
психологом, который может оценить структуру личности, психологический статус пациентов и на этой основе построить беседу;
-
педагогом, умеющим в доступной форме объяснить смысл генетического заключения.
Следует отметить, что медико-генетическое консультирование является мультидисциплинарным процессом, и подготовка врача-генетика занимает много времени. Так, например, в Европе и Америке обучение составляет 3-4 года, в нашей стране 1-2 года после окончания медицинского вуза (интернатура, ординатура). Основные составляющие медико-генетического консультирования схематично представлены на рис. 29-10.
Рис. 29-10. Основные составляющие элементы медико-генетического консультирования
Таким образом, на современном этапе развития медико-генетического консультирования наряду с основной целью профилактики врожденных пороков развития и наследственных заболеваний в семье возникает и вторая, не менее существенная цель - достижение психологического благополучия в адаптации к риску и появлению наследственной болезни в семье. Самым оптимальным вариантом функционирования медико-генетических консультаций в наше время - время новых диагностических технологий, новых подходов к лечению и профилактике наследственных и врожденных заболеваний - является согласование этих двух целей. Однако в том и другом случае путеводным принципом для врача-генетика является стремление к индивидуальному подходу в консультировании, направленному на автономию или самоопределение и персональный контроль обратившихся в медико-генетическую консультацию людей. Правомочно альтернативное поведение пациентов, направленное как на желание предупредить появление больного ребенка, так и на адаптацию к генетической болезни или риску. В любом случае необходимо стремиться находить пути к взаимному пониманию, что является ключом для дальнейшего совершенствования медико-генетического консультирования.
Этические принципы медицинской генетики сформулированы в руководстве Proposed International Guidelines on Ethical Issues in Medical Genetics and Genetic Services (15-16 декабря 1997 г., Женева), разработанном ВОЗ. В нем изложены этические принципы деятельности генетической службы как в целом, так и применительно к отдельным направлениям медицинской генетики: генетическому консультированию, генетическому скринингу, пресимптоматическому тестированию и тестированию на предрасположенность к заболеваниям, пренатальной диагностике, работе банков ДНК и генетических баз данных и др.
СПИСОК РЕКОМЕНДУЕМОЙ ЛИТЕРАТУРЫ
Козлова С.И., Демикова С.Н. Наследственные синдромы и медико-генетическое консультирование. - М.: Товарищество научных изданий КМК, 2007.
Мерфи Э.А., Чейз Г.А. Основы медико-генетического консультирования: Пер. с англ. - М.: Медицина, 1979. Стивенсон А., Дэвисон Б. Медико-генетическое консультирование: Пер. с англ. - М.: Мир, 1972.
Peter S. Harper. Practical Genetic Counseling. - 6th ed. - London: ARNOLD, 2004. Young I.D. Introduction to Risk Calculation in Genetic Counseling. - 2nd ed. - Oxford: Oxford
University Press, 1999. Young I.D. Risk estimation in genetic counseling // Emery and Rimoin?s Principles and Practice of Medical Genetics. - 4th ed. - London: Churchill Livingstone, 2002. - Р. 675-689.