Наследственные болезни : национальное руководство / Под ред. Н. П. Бочкова, Е. К. Гинтера, В. П. Пузырева - Москва : ГЭОТАР-Медиа, 2012. - 936 с. (Серия "Национальные руководства") - ISBN 978-5-9704-2231-1 |
Аннотация
Национальные руководства - первая в России серия практических руководств по основным медицинским специальностям, включающих всю основную информацию, необходимую врачу для непрерывного последипломного образования.
Национальное руководство "Наследственные болезни" содержит актуальную, современную информацию о геноме человека, общих вопросах медицинской генетики, клинической генетике. Руководство состоит из двух частей, в которых излагаются теоретические и клинические вопросы медицинской генетики. В первой части представлены новейшие данные по теоретическим вопросам медицинской генетики. Сведения об организации и функциях генома, генов и хромосом изложены в понятной для врачей форме, но без излишнего упрощения. Во второй части представлены вопросы клинической генетики, а именно методы диагностики наследственных болезней (от клинического уровня до секвенирования ДНК и РНК), принципов лечения и профилактики отдельных нозологических форм. Поскольку в национальных руководствах по другим специальностям описаны многочисленные наследственные болезни, на них можно найти ссылки [см. "Перечень наследственных болезней (синдромов), описание которых представлено в других национальных руководствах" на компакт-диске]. Приложение к руководству на компакт-диске включает более полную информацию по некоторым главам, электронную версию руководства, обширный иллюстративный материал, приложения, перечень наследственных болезней, описанных в других руководствах, фармакологический справочник. В подготовке настоящего издания в качестве авторов-составителей и рецензентов принимали участие ведущие ученые разных специальностей: генетики, иммунологи, невропатологи, фармакологи, онкологи и другие специалисты. Все рекомендации прошли этап независимого рецензирования.
Руководство предназначено для врачей-генетиков, врачей лаборантов-генетиков, врачей смежных специальностей, интернов, ординаторов, аспирантов, особенно по таким дисциплинам, как педиатрия, акушерство-гинекология, нервные болезни.
Гриф
Национальное руководство рекомендовано Российским обществом медицинских генетиков и Ассоциацией медицинских обществ по качеству
Глава 26. Принципы лечения наследственных болезней
Введение
В последние десятилетия были достигнуты значительные успехи в лечении многих наследственных заболеваний: разработаны методы ферментной заместительной терапии (ФЗТ), усовершенствованы продукты лечебного питания, появился опыт в проведении трансплантации органов, клеток и тканей. Кроме того, свои первые шаги сделала генотерапия. Все эти достижения связаны с прогрессом в области изучения механизмов патогенеза наследственных болезней и развитием новых технологий получения лекарственных препаратов, в том числе с помощью генной инженерии. Тем не менее клинический полиморфизм и низкая распространенность отдельных нозологических форм иногда не позволяют разрабатывать новые методы лечения наследственных заболеваний. Кроме того, стоимость некоторых уже созданных лекарственных препаратов очень высока, что требует особой государственной поддержки пациентов в получении современного лечения и продолжении разработки фармакологическими компаниями препаратов для небольшой группы пациентов. В конце XX в. возникли такие понятия, как «редкие заболевания» и «сиротские (орфанные) препараты». По определению, принятому в странах Евросоюза, редкими считают хронические, прогрессирующие, требующие пожизненной медицинской помощи состояния, распространенность которыми составляет менее чем 5 случаев на 10 000 населения. В России в конце 2011 г. законодательно принято определение 10 случаев на 100 000 (ФЗ № 323 от 21.11.2011 «Об основах охраны здоровья граждан Российской Федерации», ст. 44). К этой категории относятся практически все наследственные, многие онкологические заболевания, ряд инфекционных и мультифакторных болезней. Во многих странах Европы, США и Великобритании приняты законодательные акты и специальные программы, направленные на улучшение качества медицинской помощи больным с редкими заболеваниями. Статус орфанных был присвоен многим препаратам для лечения наследственных заболеваний, некоторые из которых зарегистрированы и на территории Российской Федерации (табл. 26-1).
Таблица 26-1. Примеры наследственных заболеваний, для лечения которых применяют препараты, которым присвоен статус орфанных
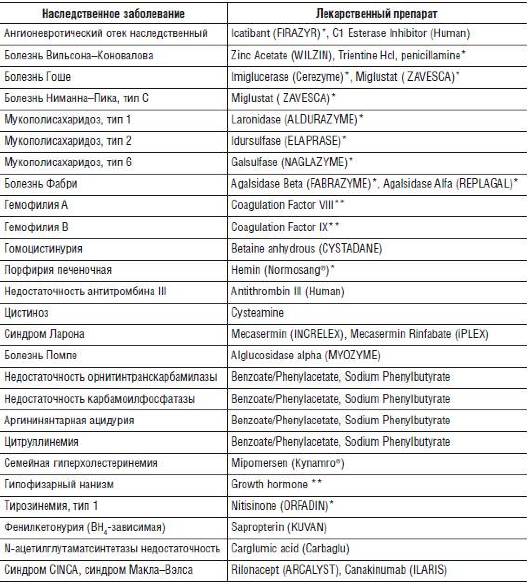
* Зарегистрированные на территории РФ препараты. ** Препараты, производимые на территории РФ.
Общие принципы лечения наследственных заболеваний
Как и при других болезнях (например, инфекционных), можно выделить три подхода к лечению наследственных заболеваний: симптоматическое, патогенетическое, этиологическое.
При симптоматическом и патогенетическом подходе используют все виды современного лечения (лекарственное, диетическое, рентгенорадиологическое, физиотерапевтическое и др.). Точный диагноз, клинические данные о состоянии больного и динамика болезни определяют тактику врача на протяжении всего периода лечения. При выборе его метода всегда необходимо взвесить риск и пользу от проводимой терапии.
Симптоматическое лечение. Для многих форм наследственных заболеваний симптоматическое лечение - единственно возможное, позволяющее улучшить качество жизни пациентов.
Примером симптоматического лечения может быть применение анальгетиков при наследственных формах мигрени, специфических транквилизаторов при психических симптомах наследственных болезней, противосудорожных препаратов при эпилепсии и др. Успехи этого раздела лечения связаны с прогрессом фармакологии, обеспечивающим все более широкий выбор лекарственных средств. Вместе с тем расшифровка патогенеза каждой болезни позволяет понять причину возникновения симптома, а на этой основе становится возможной более их тонкая лекарственная коррекция в тех случаях, когда первичное патогенетическое лечение еще невозможно. В качестве примера можно привести симптоматическое лечение мукополисахаридозов. Эти наследственные заболевания связаны с нарушением расщепления гликозаминогликанов - важнейших компонентов экстраклеточного матрикса. Несмотря на то что для некоторых форм мукополисахаридозов уже созданы препараты для ФЗТ, их применение не приводит к полной фенотипической коррекции, и пациенты нуждаются в постоянном наблюдении врачей разных специальностей и довольно разноплановом симптоматическом лечении (табл. 26-2).
Таблица 26-2. Симптоматическое лечение при мукополисахаридозах
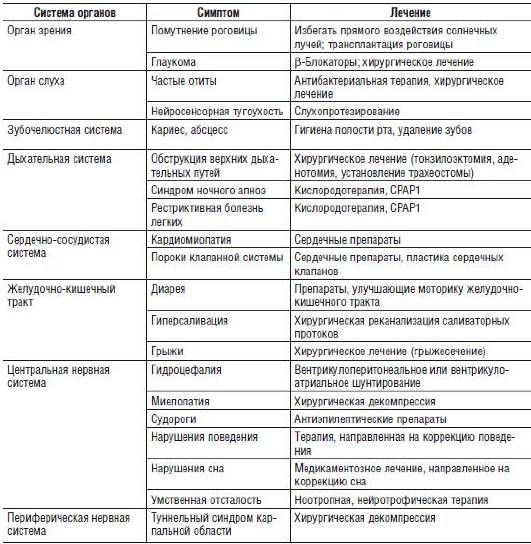
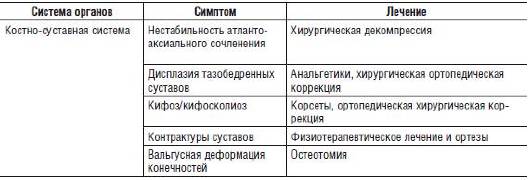
Патогенетическое лечение. На сегодняшний день число нозологических форм, для которых разработаны в разной степени эффективные методы патогенетического лечения, не превышает 150, при этом полной фенотипической коррекции можно добиться только при единичных заболеваниях. Классом наследственных болезней, для которого были достигнуты наибольшие успехи в области лечения, безусловно, считают наследственные болезни обмена веществ (НБО). Подходы к их лечению направлены на восстановление нормального гомеостаза клетки.
Несмотря на разнообразие механизмов патогенеза наследственных болезней, можно выделить несколько основных подходов к лечению (табл. 26-3).
Таблица 26-3. Основные подходы к лечению наследственных заболеваний

СНИЖЕНИЕ НАГРУЗКИ НА ПОРАЖЕННЫЙ МЕТАБОЛИЧЕСКИЙ ПУТЬ
Диетотерапия
Диетотерапия - один из основных эффективных методов лечения наследственных заболеваний, который заключается в ограничении поступления тех веществ, обмен которых нарушен в результате ферментативного блока. Эффективная диетотерапия разработана для фенилкетонурии, болезни, при которой моча приобретает запах кленового сиропа, некоторых форм гомоцистинурии и фруктоземии.
При нарушениях обмена амино- и органических кислот назначают низкобелковую диету или полусинтетические лечебные продукты, содержащие смеси незаменимых аминокислот для обеспечения возрастных потребностей ребенка в основных пищевых веществах, энергии, витаминах и минералах.
При наследственной непереносимости фруктозы исключают продукты, содержащие фруктозу и сахарозу. При этих заболеваниях можно ограничить поступление субстрата и контролировать его концентрацию.
Диетотерапия менее эффективна при тех нарушениях, когда субстрат не только поступает из пищевых источников, но и синтезируется de novo (например, при галактоземии 1-го типа). У многих пациентов с этим заболеванием, даже находящихся на строгой диете с ограничением поступления галактозы и лактозы, возникают неврологические (снижение интеллекта, нарушения речи) и эндокринные нарушения.
Хотя для ряда НБО разработана диетотерапия, ее эффект значительно ниже теоретически ожидаемого. Это касается Х-сцепленной адренолейкодистрофии (масло Лоренцио), синдрома Смита-Лемли-Опитца (высокохолестериновая диета) и многих других заболеваний (табл. 26-4).
Таблица 26-4. Диетотерапия при наследственных заболеваниях
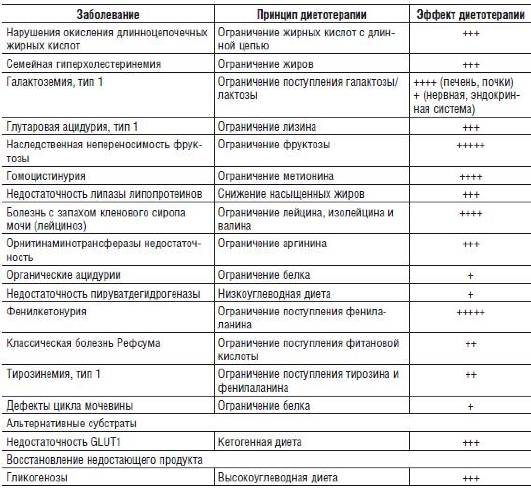
Ингибирование ферментов, находящихся выше метаболического блока
Использование NTBC [2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione] для лечения тирозинемии 1-го типа - один из примеров эффективного применения этого подхода. NTBC ингибирует 4-гидроксифенилпируватдегидрогеназу - фермент, расположенный выше генетически обусловленного блока при тирозинемии 1-го типа (фумарилацетоацетазы), что предотвращает продукцию фумарилацетоацетата и сукцинилацетона - основных токсичных веществ, образующихся при этом заболевании (рис. 26-1).
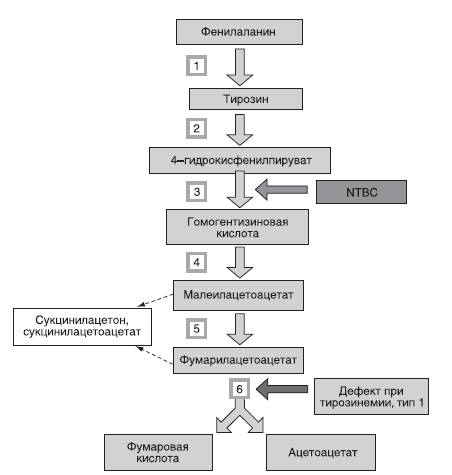
Рис. 26-1. Метаболизм тирозина, принцип лечения тирозинемии, тип 1: 1 - система гидроксилирования фенилаланина; 2 - тирозинтрансаминаза; 3 - 4-гидроксифенилпируват-диоксигеназа; 4 - гомогентизатоксидаза; 5 - малеилацетоацетат изомераза; 6 - фумарилацетоацетаза
Сравнительно недавно сходный подход стали применять для лечения лизосомных болезней накопления (ЛБН). Большинство известных на сегодняшний день групп ЛБН связано с нарушением определенных стадий катаболизма макромолекул (мукополисахаридозы, гликосфинголипидозы, гликопротеинозы и др.), и основные патогенетические механизмы обусловлены накоплением в клетках нерасщепленных субстратов. Примерно три десятилетия назад был предложен оригинальный подход к лечению ЛБН - снижение накопления субстратов за счет ограничения их биосинтеза. Особое внимание привлекла группа гликосфинголипидозов. Заболевания этой группы обусловлены нарушением различных реакций расщепления сложных молекул - гликосфинголипидов. Известно, что последние синтезируются из общего предшественника - церамида, а пути их расщепления различаются. Принцип лечения заключается в следующем: обратимые ингибиторы ключевой стадии биосинтеза (превращение церамида в глюкозилцерамид) позволяют снизить синтез гликосфинголипидов до уровня, с которым может справиться мутантный фермент, отвечающий за определенную реакцию расщепления этих биологических молекул (рис. 26-2). В результате происходит уменьшение концентрации гликосфинголипидов, накапливаемых в лизосомах.
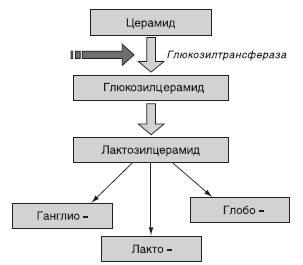
Рис. 26-2. Синтез гликосфинголипидов. Стрелкой обозначено место действия ингибиторов фермента при субстратредуцирующей терапии
Ингибирование церамидглюкозилтрансферазы можно осуществить с помощью небольших молекул иминосахаров. Механизм их действия до конца не выяснен, но хорошо объясняется структурной схожестью между иминосахарами и церамидом. Другой класс соединений, ингибирующих эту же стадию биохимического пути биосинтеза, - морфолино- и пирролидиновые производные (PDMP-series). Одно из этих соединений - N-butyldeoxynojirimycin (NB-DNJ, miglustat) - зарегистрировано как лекарственный препарат, и его уже применяют в клинической практике для лечения болезни Гоше 1-го типа и болезни Ниманна-Пика, тип С.
Существенное преимущество этих препаратов - их возможность проникать через ГЭБ, что позволяет применять их для лечения неврологических форм заболеваний. Тем не менее этот подход применим только для случаев, когда сохраняется высокая остаточная активность фермента. Это наиболее характерно для поздних, более доброкачественных форм заболеваний. Работы в этом направлении продолжаются, и, возможно, будут созданы лекарственные средства для лечения и других форм ЛБН.
КОРРЕКЦИЯ НЕДОСТАТКА ПРОДУКТА БЛОКИРОВАННОЙ РЕАКЦИИ
Восполнение недостающего продукта
Если основные механизмы патогенеза заболевания связаны преимущественно с недостаточностью продукта блокированной ферментной реакции, то его восполнение крайне эффективно для купирования основных клинических симптомов болезни. Этот подход с успехом применяют для лечения гликогенозов, дефектов метаболизма креатинина, биоптерина и нарушений синтеза стероидных гормонов.
Увеличение поступления субстрата
Фармакологическое повышение концентрации субстрата может быть эффективным при нарушениях мембранных транспортных белков: например, при дефектах карнитинового транспортера применяют высокие концентрации карнитина, при ННН-синдроме (гипераммониемия, гиперорнитинемия, гомоцитрулинемия) - орнитин. Безусловно, такое лечение возможно только в тех случаях, когда субстрат в высоких концентрациях не токсичен.
Введение альтернативных субстратов
Компенсировать нарушение работы определенного метаболического пути можно за счет увеличения нагрузки на другие, сходные по своим конечным продуктам биохимические реакции. Например, при мутационном повреждении белка - переносчика GLUT1, который обеспечивает транспорт глюкозы через ГЭБ, возникает недостаток глюкозы в клетках ЦНС. Кетогенная диета при этом заболевании обеспечивает альтернативное «топливо» для нервной системы, что позволяет купировать эпилептические приступы, ведущие в клинической картине этого нарушения. Другими примерами такого лечения служит применение среднецепочечных триглицеридов при нарушениях β-окисления длинноцепочечных жирных кислот.
СНИЖЕНИЕ МЕТАБОЛИЧЕСКОЙ ТОКСИЧНОСТИ НАКАПЛИВАЕМЫХ ПРОДУКТОВ
Выведение токсичных метаболитов
Применение препаратов, связывающих и переводящих в нетоксичную форму различные метаболиты, широко используют в качестве самостоятельного лечения и в комбинации с другими видами терапии при НБО.
Левокарнитин (L-карнитин) образует с органическими кислотами ацилкарнитиновые эфиры, которые менее токсичны и лучше выводятся почками, что позволяет применять его при различных органических ацидуриях . Сходными свойствами обладает аминокислота глицин, которая образует глициновые конъюгаты с некоторыми из органических кислот (например, изовалериановой).
При дефектах цикла мочевины применяют бензоат натрия и фенилбутират натрия, которые выводят избыток ионов аммония из организма.
Снижение токсичности метаболитов
Для лечения заболеваний, клинические признаки которых обусловлены действием избытка метаболитов на рецепторы, одним из возможных подходов считают блокаду агонистами накапливаемого метаболита. Например, применение кетамина - агониста N-метил-D-аспартатных рецепторов (NMDA-рецепторов) уменьшает возбуждающий эффект глицина на NMDA-рецептор при некетотической гиперглицинемии.
Интересный подход, разработанный для лечения фенилкетонурии, - применение больших нейтральных L-аминокислот (LNAA), которые не позволяют фенилаланину проникать через ГЭБ. Известно, что LNAA и фенилаланин транспортируются через ГЭБ одним переносчиком, который имеет разное сродство к этим аминокислотам. В ряде работ было показано, что если в крови сохраняется высокая концентрация LNAA, то они преимущественно переносятся из крови через ГЭБ, а содержание фенилаланина в клетках нервной системы не повышается, хотя в крови остается высоким. Добавки LNAA в определенной концентрации позволяют пациентам с фенилкетонурией не придерживаться очень строгой диеты.
Активация альтернативного метаболического пути. Оригинальный подход к лечению наследственных заболеваний - активация минорных метаболических путей, позоляющая переводить токсичные метаболиты в более безопасные соединения. Классический пример подобного подхода к лечению - применение бетаина при гомоцистинурии. Бетаин участвует в реакции метилирования гомоцистеина, катализируемой бетаингомоцистеинметилтрансферазой. В результате применения бетаина концентрация гомоцистеина снижается, что приводит к улучшению клинической симптоматики (рис. 26-3).
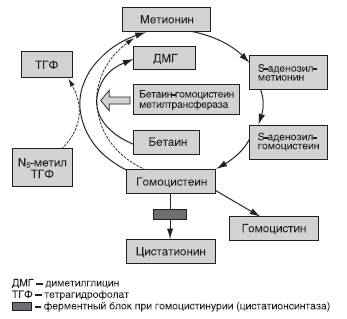
Рис. 26-3. Метаболизм метионина, принцип лечения гомоцистинурии
СТИМУЛЯЦИЯ РАБОТЫ ДЕФЕКТНОГО ФЕРМЕНТА
Применение коферментов
Существует множество заболеваний, которые обусловлены изменениями, затрагивающими участки фермента, критичные для связывания с кофактором, или вызваны нарушениями метаболизма самих кофакторов. При этих состояниях применение высоких доз витаминов позволяет добиться значительного клинического улучшения.
Наиболее яркий пример такого подхода - лечение недостаточности биотинидазы. При этом заболевании снижается активность фермента, который расщепляет биоцитин на свободный биотин (витамин Н) и лизин. Нарушение функции карбоксилаз, для которых биотин служит ковалентно связанным кофактором, приводит к накоплению субстратов, контролируемых карбоксилазами ферментных реакций. Эти субстраты и их производные (β-гидроксиизовалериановая, молочная, β-гидроксипропионовая, фумаровая, 2-оксиглутаровая и метиллимонная кислоты) оказывают токсическое действие на ЦНС и другие ткани, приводя к развитию метаболического ацидоза и вторичной гипераммониемии. Применение высоких доз биотина позволяет достичь стойкого клинического эффекта и приводит к полному купированию всех симптомов заболевания.
Для многих болезней из группы органических ацидурий и аминоацидопатий описаны особые витаминчувствительные формы, которые хорошо поддаются лечению витаминами в больших дозах (B12-зависимая метилмалоновая ацидурия, В6-чувствительная гомоцистинурия; табл. 26-5). Следует отметить, что назначение коферментов позволяет стабилизировать фермент и повысить его остаточную активность даже при классических формах заболеваний. Примером этого служит применение тетрагидробиоптеринаρ (ВН4) при классической фенилкетонурии. В ряде работ было убедительно продемонстрировано, что некоторые мутации в гене фенилаланингидроксилазы нарушают конформацию и стабильность фермента, а применение ВН4 позволяет достичь значительного повышения его активности, что дает возможность больным не придерживаться строгой диеты.
Таблица 26-5. Лечение заболеваний, для которых описаны кофакторзависимые формы
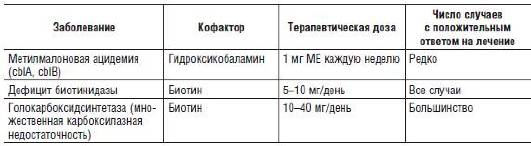
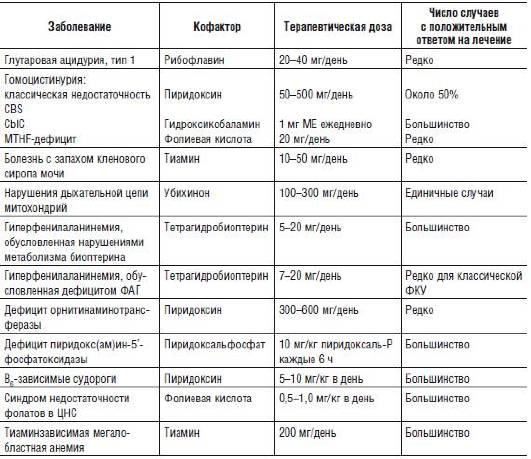
Примечание. Cbl - кобаламин, CBS - цистотионин-β-синтетаза, ФАГ - фенилаланингидроксилаза, MTHF - метилентетрагидрофолатредуктаза.
Следует отметить, что при определенном заболевании необходимо использовать соответствующую форму кофермента. Например, пиридоксальфосфат, а не пиридоксинρ, эффективен при недостаточности пиридокс(ам)ин-5′-фосфатоксидазы. Несмотря на то что активными коферментами для ряда ферментов (метилмалонил-КоА-мутазы и S-аденозилметилтрансферазы) служат аденозилкобаламин и метилкобаламин, только гидроксикобаламинρ эффективно транспортируется в клетки, и, следовательно, его применяют в лечении кобаламинзависимых нарушений.
?- Ферментиндуцирующая терапия. Фармакологические шапероны.
Этот особый подход предложен для лечения наследственных заболеваний, при которых нарушаются сворачивание (фолдинг) белка и его доставка в определенный компартмент клетки.
Чтобы стать узнаваемым для ферментов, белков-переносчиков и других молекул, белок должен обладать определенной пространственной конфигурацией. Образование аномального по структуре белка запускает серию реакций по его утилизации, и белок разрушается при участии особой убиквитиновой системы клетки, даже не достигнув основного пункта своего назначения (например, лизосом).
Ряд мутаций в генах приводит к нарушениям фолдинга белка и вызывает либо его накопление, либо быстрое расщепление. Это, как правило, миссенс-мутации и небольшие делеции без сдвига рамки считывания, которые не затрагивают функционально значимые домены белка (активные центры, рецепторсвязывающие сайты). Такие белки сохранили бы функциональную активность, если бы достигли места своего назначения. Классический пример нарушений фолдинга белка - мутация ΔF508 в гене CFTR (муковисцидоз). Она приводит к тому, что белок не проходит необходимые стадии созревания в эндоплазматическом ретикулуме. В результате он не транспортируется в плазматическую мембрану клетки, а остается в последнем, где подвергается расщеплению.
Недостаточность α1-антитрипсина и некоторые лизосомные болезни накопления (болезнь Фабри, GM1-ганглиозидоз, болезнь Гоше) обусловлены мутациями, которые приводят к «застреванию» белков на полпути и отсутствию транспортировки в нужный компартмент клетки. Давно известно, что некоторые соединения могут служить стабилизаторами белков и помогают им образовывать более устойчивую конформацию. Эти вещества называют химическими шаперонами по аналогии с белками-шаперонами, которые принимают участие в поддержании третичной структуры и доставке синтезированных белков к клетке. К простейшим химическим шаперонам относят глицерин, сахарозу, галактозу и др. Химические шапероны неспецифичны, а их применение в клинических целях ограничено, поскольку для достижения эффекта требуются высокие концентрации, создание которых приводит к возникновению множества побочных эффектов. Для прицельной стабилизации ферментов и других белков были предложены молекулы, которые назвали фармакологическими шаперонами. Это низкомолекулярные, обычно гидрофобные молекулы, которые служат аналогами субстратов, обратимых ингибиторов ферментов и кофакторов или агонистами рецепторов и стабилизируют специфичные для них белки.
В ряде эксперментов in vitro было показано, что некоторые соединения, связываясь с каталитическим центром фермента, могут стабилизировать его конформацию. В работах на культуре клеток с мутациями в гене α-галактозидазы (болезнь Фабри) было установлено, что добавление 1-deoxygalactonojirimycinρ в культуральную среду в концентрациях, близких к ингибиторным, может стабилизировать мутационно измененный фермент. Введение препарата трансгенным мышам, имеющим дефект гена α-галактозидазы, приводит к повышению активности фермента во многих тканях, прежде всего в сердечной мышце, селезенке и почках. Это позволяет применять этот подход для большого числа заболеваний, обусловленных миссенс-мутациями. Показано, что N-(n nonyl)deoxynojirimycinρ (NN-DNJ) приводит к увеличению активности мутантного фермента в культуре кожных фибробластов при болезни Гоше (мутация N370S). В ряде работ показана эффективность применения этого метода для коррекции GM1-ганглиозидоза. Фармакологические шапероны для лечения ЛБН уже проходят завершающую стадию клинических испытаний.
ВОССТАНОВЛЕНИЕ НЕДОСТАЮЩЕГО ФЕРМЕНТА
Ферментная заместительная терапия
ФЗТ при заболеваниях, обусловленных недостаточностью определенного белка-фермента, - один из самых надежных и физиологичных методов лечения. Наибольшие успехи в ее применении были достигнуты для ЛБН. Эта группа наследственных моногенных заболеваний включает более 40 различных форм, большинство из которых связано с нарушениями функции определенного фермента лизосом.
Идея о возможности коррекции ЛБН путем введения фермента была впервые высказана еще де Дювом в 1964 г., но до внедрения этого метода лечения в клиническую практику прошло еще около 25 лет. Важнейшей предпосылкой для разработки ФЗТ послужило открытие механизмов посттрансляционной модификации и транспорта лизосомных ферментов. В начале 70-х гг. XX в. было показано, что на поверхности клеточных мембран находятся маннозо-6-фосфатные рецепторы, которые могут связывать и переносить фермент внутрь клетки.
В экспериментах на культуре клеток с низкой активностью лизосомных ферментов было обнаружено, что внесенный в культуральную среду экзогенный фермент способен проникать в клетку и успешно катаболизировать накопленный внутриклеточный субстрат. При этом было показано, что даже поддержания 1-5% нормальной активности фермента достаточно для коррекции метаболического дефекта.
В начале 70-х гг. пилотные клинические исследования проводили для терапии болезни Зандгоффа, Помпе, Фабри и Гоше. В каждом случае очищенный человеческий фермент демонстрировал свою активность in vitrо, и накапливаемые метаболиты выводили из клеток. В клинических испытаниях было показано, что внутренние органы хорошо отвечают на лечение, но при ЛБН с поражением ЦНС не удавалось устранить неврологические нарушения, поскольку фермент не проникает через ГЭБ. Все попытки повысить его проницаемость также оказались малоуспешными. Именно поэтому основные разработки ФЗТ были направлены на создание препаратов для лечения заболеваний без вовлечения ЦНС.
В начале 90-х гг. были опубликованы данные о том, что ФЗТ высокими дозами модифицированного фермента глюкоцереброзидазы, выделенного из плаценты человека, приводит к значительному клиническому улучшению у пациентов с болезнью Гоше 1-го типа.
Успех этого метода лечения стимулировал ученых к разработке ФЗТ для других форм ЛБН (табл. 26-6). В дальнейшем этому способствовало совершенствование генно-инженерных методов - создание эукариотических систем, способных экспрессировать большие количества рекомбинантного человеческого фермента, и линий нокаутных мышей, на которых можно выполнять доклинические исследования ФЗТ.
Таблица 26-6. Ферментная заместительная терапия при лизосомных болезнях накопления
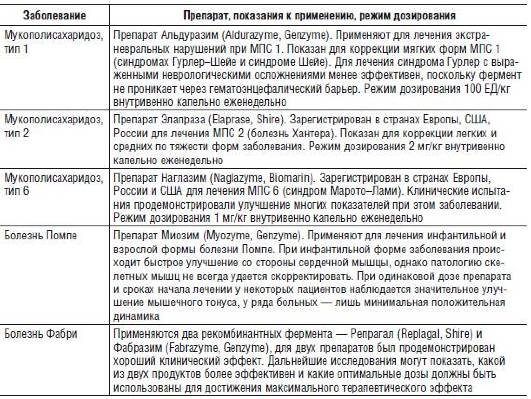

Известно, что большинство белков человека можно синтезировать с помощью прокариотических клеток, но этот подход неприемлем для лизосомных ферментов, нуждающихся в особых посттрансляционных модификациях (N-гликозилирование), которые не происходят у прокариот. Именно поэтому в качестве первых систем для производства лизосомных ферментов человека были выбраны клетки яичников китайского хомячка. Они легко поддаются культивированию, и в них реализуется очень близкая к человеческой система посттрансляционной модификации белков. Сверхэкспрессия ДНК лизосомных ферментов в них приводит к тому, что рекомбинантный человеческий белок секретируется в культуральную среду, а это позволяет получать большое количество фермента. Другие модели, такие как культура человеческих фибробластов, трансгенные животные и даже растения, также применяют для наработки терапевтических ферментов.
На сегодняшний день препараты для ФЗТ получены для лечения болезни Гоше, Помпе, мукополисахаридоза 1-го, 2-го и 6-го типов, а также болезни Фабри. Клинические испытания показали, что ФЗТ безопасна, не вызывает выраженных побочных эффектов и приводит к выведению негидролизованного субстрата. Кроме того, ее хорошо переносят больные. Реакции на введение препарата связаны с образованием антител против белка, но они не постоянны и их можно купировать стандартными средствами.
Основное ограничение ФЗТ - неспособность рекомбинантного фермента проникать через ГЭБ, что не позволяет применять этот метод для лечения неврологических заболеваний. Один из подходов к решению данной проблемы - введение препарата в спинномозговой канал. В экспериментах, проведенных на моделях животных с мукополисахаридозом 1-го типа, было показано, что фермент может достичь нейронов и клеток глии, но этот метод его введения вряд ли получит широкое распространение. Существуют также трудности с коррекцией патологических изменений костно-суставной системы.
Ряд исследователей предпринимают попытки решить задачу доставки фермента к клеткам соединительной ткани, что особенно актуально для лечения мукополисахаридозов (в частности, 4-го типа), но пока они малоуспешны. Препараты для ФЗТ вводят внутривенно несколько раз в месяц. Поскольку такое лечение проводят пожизненно, неизбежно возникает затруднение венозного доступа вследствие изменений стенок сосудов. На животных моделях сейчас разрабатывают и другие способы введения недостающего фермента. Один из них - микроинкапсулирование: генетически модифицированные клетки, продуцирующие необходимый белок, погружают в иммунозащитные микрокапсулы и имплантируют в организм пациента (подкожно или внутрибрюшинно). Такие депо могут долгое время вырабатывать недостающие организму ферменты или гормоны.
ТРАНСПЛАНТАЦИЯ ГЕМОПОЭТИЧЕСКИХ СТВОЛОВЫХ КЛЕТОК
Трансплантацию гемопоэтических стволовых клеток (ТГСК) довольно широко применяют при наследственных заболеваниях: нарушениях иммунной системы, анемиях, некоторых болезнях обмена, ЛБН и пероксисомных заболеваниях (см. гл. 22). Оптимальный аллогенный донор для ТГСК - брат или сестра больного, имеющие идентичный человеческий лейкоцитарный антиген (HLA). Если среди членов семьи нет HLA-идентичного донора, то в некоторых случаях, главным образом у молодых пациентов, можно выполнить ТГСК с использованием костного мозга неродственного донора, совместимого по HLA. Полиморфность системы HLA настолько велика, что далеко не всегда удается найти подходящего донора. Результативность трансплантации зависит от генотипа, статуса донора (гетерозигота или здоровый донор), возраста пациента на момент выполнения трансплантации, степени выраженности органных поражений на момент проведения ТГСК, функционирования донорского органа и посттрансплантационной реабилитации.
Первое заболевание из группы ЛБН, при котором была проведена ТГСК, - мукополисахаридоз 1-го типа (синдром Гурлер). Очевидно, что ТГСК при этом состоянии приводит к значительному улучшению состояния пациентов, хотя некоторые осложнения не поддаются полной коррекции. Кроме того, для предотвращения неврологических нарушений ТГСК следует провести как можно раньше (в идеале - до 24-го месяца жизни). Высокий риск процедуры, трудности подбора донора, а также посттрансплантационные осложнения создают значительные проблемы в применении этого метода лечения. ТГСК при синдроме Гурлер в настоящее время - стандарт лечения этого заболевания. Проведение трансплантации в раннем возрасте приводит к полной коррекции висцеральных нарушений (гепатоспленомегалия), кардиомиопатии и легочных изменений, а также к значительному улучшению неврологического статуса.
При лечении других ЛБН роль ТГСК менее ясна. Возможно, она даже противопоказана пациентам с крайне тяжелыми нейродегенеративными заболеваниями (например, синдром Санфилиппо, мукополисахаридоз 3-го типа). На доклинической стадии ТГСК может быть эффективна при болезни Краббе и метахроматической лейкодистрофии . Одно из перспективных направлений - применение других стволовых клеток (например, мезенхимальных стволовых клеток костного мозга), а также сочетание ТГСК с другими методами лечения.
ТРАНСПЛАНТАЦИЯ ОРГАНОВ
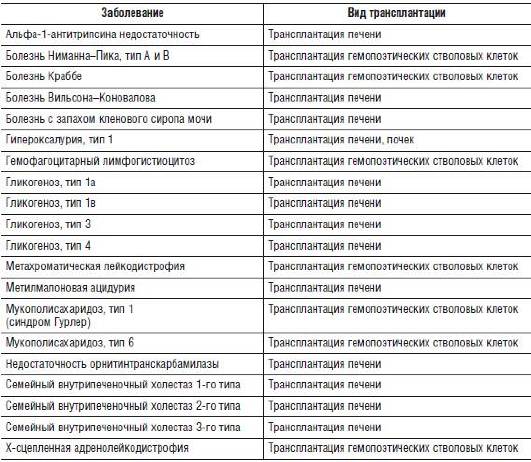
Трансплантацию печени или гепатоцитов успешно применяют при лечении заболеваний из группы нарушений цикла мочевины, некоторых органических ацидурий и тяжелых форм гликогенозов. В то время как определены четкие показания для трансплантации при гликогенозе 4-го типа или печеночной недостаточности при болезни Вильсона-Коновалова, показания для других врожденных нарушений не определены и процесс принятия решения часто очень сложен. Смертность, связанная с пересадкой печени, очень высока у пациентов с тяжелыми нарушениями межуточного обмена. Более того, трансплантация при органических ацидуриях не обязательно уменьшает риск возникновения осложнений, связанных с заболеванием. Тем не менее для подобных состояний с плохим прогнозом при использовании обычного медикаментозного лечения пересадка печени может быть эффективным методом.
Трансплантацию почек применяют для лечения терминальной стадии почечной недостаточности при таких НБО, как цистиноз и болезнь Фабри. Сочетанную пересадку печени и почки проводят при метилмалоновой ацидемии и первичной гипероксалурии 1-го типа.
Наследственные заболевания, при которых применяют трансплантацию печени и других органов, приведены в табл. 26-7.
Таблица 26-7. Трансплантация органов и тканей при наследственных заболеваниях (примеры заболеваний)
ГЕНОТЕРАПИЯ
Успехи генотерапии на фоне достижений биохимической коррекции наследственных болезней выглядят гораздо скромнее. Протоколы лечения разработаны только для единичных наследственных заболеваний. Основные принципы генотерапии подробно изложены в гл. 22.
Заключение
Лечение наследственных болезней - необычайно трудная задача, не всегда эффективно решаемая. Крайне важным в их лечении считают постоянное наблюдение пациента многими специалистами и образование родителей, поскольку от того, как семья воспринимает процесс лечения, насколько родители понимают необходимость, например, диетотерапии и осведомлены о возможных побочных эффектах препаратов, зависит и результативность лечения. В ряде случаев семьям приходится принимать очень непростые решения, которые касаются таких процедур, сопряженных с высоким риском развития осложнений, как трансплантация органов и тканей. Нестойкость, а часто и недостаточная выраженность эффектов лечения не снимают вопроса о ее постоянном проведении не только с клинической точки зрения, но и по деонтологическим соображениям.
Определенный прогресс в лечении наследственных болезней уже произошел, но он, безусловно, лишь частичен. Для восстановления нормального гомеостаза необходимо развивать методы генотерапии, пересадки органов и тканей, фармакотерапии и улучшения функционирования поддерживающих систем. Последние достижения молекулярной биологии, возможности генной инженерии, фармакологии и биохимии позволяют надеяться, что могут быть созданы принципиально новые методы лечения наследственных болезней.
СПИСОК РЕКОМЕНДУЕМОЙ ЛИТЕРАТУРЫ
Бочков Н.П., Пузырев В.П., Смирнихина С.А. Клиническая генетика. - М.: ГЭОТАРМедиа, 2010. - 554 с.
Бочков Н.П. и др. Клеточная терапия наследственных болезней // Вестн. РАМН. - 2008. - № 10. - С. 20-28.
Гинтер Е.К. Медицинская генетика. - М.: Медицина, 2003. - 448 с.
Горбунова В.Н., Баранов В.С. Введение в молекулярную диагностику и генотерапию наследственных заболеваний. - СПб., 1997. - 287 с.
Hoffmann G.F., Zschocke J., William L. Inherited Metabolic Diseases: A Clinical Approach. - Springer, 2010.
Leiden J.M. Human gene therapy: the good, the bad, and the ugly // Circ. Res. - 2000. - Vol. 86. - P. 923-925.