Наследственные болезни : национальное руководство / Под ред. Н. П. Бочкова, Е. К. Гинтера, В. П. Пузырева - Москва : ГЭОТАР-Медиа, 2012. - 936 с. (Серия "Национальные руководства") - ISBN 978-5-9704-2231-1 |
Аннотация
Национальные руководства - первая в России серия практических руководств по основным медицинским специальностям, включающих всю основную информацию, необходимую врачу для непрерывного последипломного образования.
Национальное руководство "Наследственные болезни" содержит актуальную, современную информацию о геноме человека, общих вопросах медицинской генетики, клинической генетике. Руководство состоит из двух частей, в которых излагаются теоретические и клинические вопросы медицинской генетики. В первой части представлены новейшие данные по теоретическим вопросам медицинской генетики. Сведения об организации и функциях генома, генов и хромосом изложены в понятной для врачей форме, но без излишнего упрощения. Во второй части представлены вопросы клинической генетики, а именно методы диагностики наследственных болезней (от клинического уровня до секвенирования ДНК и РНК), принципов лечения и профилактики отдельных нозологических форм. Поскольку в национальных руководствах по другим специальностям описаны многочисленные наследственные болезни, на них можно найти ссылки [см. "Перечень наследственных болезней (синдромов), описание которых представлено в других национальных руководствах" на компакт-диске]. Приложение к руководству на компакт-диске включает более полную информацию по некоторым главам, электронную версию руководства, обширный иллюстративный материал, приложения, перечень наследственных болезней, описанных в других руководствах, фармакологический справочник. В подготовке настоящего издания в качестве авторов-составителей и рецензентов принимали участие ведущие ученые разных специальностей: генетики, иммунологи, невропатологи, фармакологи, онкологи и другие специалисты. Все рекомендации прошли этап независимого рецензирования.
Руководство предназначено для врачей-генетиков, врачей лаборантов-генетиков, врачей смежных специальностей, интернов, ординаторов, аспирантов, особенно по таким дисциплинам, как педиатрия, акушерство-гинекология, нервные болезни.
Гриф
Национальное руководство рекомендовано Российским обществом медицинских генетиков и Ассоциацией медицинских обществ по качеству
Глава 5. Моногенное наследование признаков у человека
Введение
Основными понятиями при рассмотрении менделевского наследования признаков и болезней у человека являются «фенотип» и «генотип». Под термином фенотип принято понимать сумму всех внешних характеристик человека. Следует отметить, что когда говорится о внешних характеристиках, то следует подразумевать не только такие внешние признаки, как рост, цвет глаз или число пальцев на руках и ногах, но и различные физиологические, биохимические и даже молекулярные характеристики, которые могут измениться в результате действия генов. Фенотипические признаки, с которыми сталкивается медицинская генетика, - это наследственные болезни и их симптомы.
Под термином генотип понимают сумму всех генов организма. Однако нередко понятие «генотип» используют для описания состояния отдельного или нескольких генов. Генотип в значительной мере определяет фенотип, а гены - отдельные фенотипические признаки.
Фенотип в отличие от генотипа может меняться в течение жизни, генотип при этом остается постоянным, свидетельство этому наш собственный онтогенез. В течение жизни внешне человек меняется, старея, а генотип - нет. За одним и тем же фенотипом могут стоять разные генотипы и, напротив, при одном и том же генотипе фенотипы могут различаться. Последнее утверждение подкреплено изучением монозиготных близнецов. Их генотипы идентичны, а фенотипически они могут различаться по массе тела, росту, поведению и т.д. Из этого следует, что не существует в абсолютном большинстве случаев однозначной связи между генами и фенотипическими признаками, и проявление генов может изменяться как под действием факторов окружающей среды, так и в результате взаимодействия с другими генами. Очевидно, что между симптомами наследственного заболевания, такими как: отсутствие ушной раковины, судороги, умственная отсталость, кисты в почках и многими другими, и изменением одного белка в результате мутации в каком-то конкретном гене - дистанция огромного размера. Мутантный белок - продукт мутантного гена - должен каким-то образом взаимодействовать с сотнями, если не с тысячами других белков, кодируемых другими генами, чтобы в результате изменился какой-то нормальный признак или появился патологический. Кроме того, продукты генов, принимающих участие в становлении любого фенотипического признака, могут взаимодействовать и модифицироваться факторами окружающей среды. Вместе с тем, когда речь идет о моногенно наследуемых признаках (например, наследственных болезнях), то действие мутантного гена в этом случае не затушевывается многочисленными взаимодействиями его продукта с продуктами других генов или с факторами окружающей среды, и наличие мутантного гена(-ов) определяет изменение фенотипа. Это происходит как в сложном механизме, когда дефекта одной детали достаточно, чтобы при тысячах других нормальных деталях механизм не работал.
Моногенное наследование признаков у человека называют еще менделевским, так как правила, согласно которым оно происходит, установил Грегор Мендель еще в 1865 г. Ниже приведены правила, установленные Менделем в ходе выполненных им опытов.
Первое правило называется правилом доминирования, и его суть сводится к тому, что из двух копий каждого гена, называемых аллелями и содержащихся в каждой клетке организма, одна может подавлять или, правильнее, маскировать проявление второй копии (аллеля). В тех случаях, когда аллели гена одинаковы, особь с таким генотипом называют гомозиготной, а когда они разные - гетерозиготной. Доминантный аллель, следовательно, определяет характер признака, даже находясь в гетерозиготном состоянии, а рецессивный аллель определяет характер признака только тогда, когда он находится в гомозиготном состоянии. Соответственно все менделирующие признаки, включая наследственные болезни, делятся на доминантные и рецессивные. Если у гетерозиготной особи проявляются оба аллеля, т.е. нет доминирования одного аллеля над другим, то такие аллели называют кодоминантными. Хорошо известным примером кодоминирования являются аллели А и В группы крови АВ0. У людей с IV группой крови проявляются как А-, так и В-антигены.
Мендель также предположил (теперь это доказано), что в половые клетки родителей случайным образом попадает один из аллелей каждого гена, так что одна половина гамет несет один аллель, а вторая - другой. Это утверждение называется вторым правилом Менделя, или правилом расщепления. Если оба родителя являются гетерозиготами по какому-то гену, то в потомстве таких родителей будет наблюдаться расщепление, так что 3/4 потомков будут иметь доминантный признак и только 1/4 рецессивный, что обусловлено случайным объединением гамет родителей, несущих разные аллели гена (следует отметить, что расщепление по генотипу будет иным, а именно 1:2:1). Соответственно, когда только один родитель гетерозиготен, а второй гомозиготен по рецессивному гену, то расщепление по наличию доминантного и рецессивного признаков будет 1:1. Если один родитель будет гомозиготен, а второй - гетерозиготен по доминантному гену, то фенотипически все потомство будет иметь только доминантный признак. Для понимания того, как действует правило расщепления, лучше всего воспользоваться решеткой Пеннета, которую этот английский генетик предложил для графического представления результатов различных скрещиваний (табл. 5-1, 5-2, 5-3).
Таблица 5-1. Решетка Пеннета, иллюстрирующая результаты расщепления в потомстве от брака двух гетерозиготных родителей
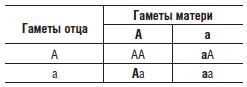
Таблица 5-2. Решетка Пеннета, иллюстрирующая результаты расщепления в потомстве от брака родителей, один из которых гетерозиготен, а второй гомозиготен по рецессивному гену

Таблица 5-3. Решетка Пеннета, иллюстрирующая результаты расщепления в потомстве от брака родителей, один из которых гетерозиготен, а второй гомозиготен по доминантному гену
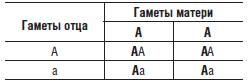
Менделю было ясно, что наблюдаемые расщепления в потомстве от скрещиваний родителей с разными генотипами являются событиями вероятностными, и их можно выявить только на большом числе потомков. Из теории вероятностей следуют два правила - правило умножения и правило сложения вероятностей.
Правило умножения гласит, что если какие-то события наблюдаются независимо друг от друга, то вероятность того, что два события будут наблюдаться одновременно, равна произведению вероятностей этих событий. Вероятность образования гамет с рецессивным геном у родителей, гетерозиготных по этому гену, составляет 1 /2 для каждого родителя. Вероятность встречи таких гамет с рецессивным геном при образовании зигот будет равна произведению вероятностей образования таких гамет у каждого из родителей, т.е. 1/2 ×1/2 =1/4.
Правило сложения гласит: если необходимо узнать вероятность реализации либо одного, либо другого события, то вероятности каждого из этих событий складываются. Так, если необходимо вычислить вероятность гомозиготного потомства в браке гетерозиготных родителей, то нужно сложить вероятности рецессивных и доминантных гомозигот, т.е. 1/4 +1/4 =1/2.
Этими правилами приходится довольно часто пользоваться врачам-генетикам во время медико-генетического консультирования при расчете вероятностей тех или иных событий в семьях, имеющих ребенка с наследственным заболеванием.
Третье правило Менделя, или правило независимого комбинирования, которое формулируется так: гены, определяющие различные признаки, наследуются независимо друг от друга. Это правило относится не к наследованию альтернативных состояний одного признака, а к двум и большему числу признаков. Знание этого правила будет иметь значение при рассмотрении картирования генов с помощью анализа сцепления.
Ниже приведено расщепление в потомстве от брака родителей, гетерозиготных по двум генам одновременно (АаВв), причем каждый из этих генов влияет на разные признаки. Проще всего это сделать, использовав решетку Пеннета (табл. 5-4).
Таблица 5-4. Решетка Пеннета, иллюстрирующая результаты расщепления в потомстве от брака родителей, гетерозиготных по двум генам
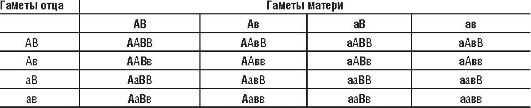
Как следует из табл. 5-4, в потомстве от брака двойных гетерозигот наблюдается 4 фенотипа:
Соотношение между этими фенотипами в том порядке, как они записаны выше, составляет 9:3:3:1. Эти соотношения легко получить, перемножая вероятности соответствующих фенотипов при моногибридном расщеплении. Так, вероятность доминантного фенотипа для каждого признака в моногибридном скрещивании составляет 3/4. При их независимости друг от друга вероятность их совместного проявления будет равна 3/4 ×3/4 =9/16. Следует подчеркнуть, что соотношение генотипов при дигибридном скрещивании иное, чем соотношение фенотипов: 1ААВВ:2АаВВ:2ААВв:4АаВв:1ААвв:2Аавв:1ааВВ:2ааВв:1аавв.
Особенности проявления менделевских правил наследования у человека
У человека, по понятным причинам, невозможны контролируемые скрещивания. Кроме того, число детей в семье, как правило, бывает небольшим. Для того чтобы доказать аутосомно-доминантный или аутосомно-рецессивный характер наследования заболевания, необходимо собрать достаточно большое число семей с большим числом детей в них. Если учесть, что абсолютное большинство наследственных заболеваний встречается редко, то становится ясно, что чаще всего медицинскому генетику приходится пользоваться упрощенными требованиями к доказательствам определенного типа наследования. Эти упрощенные требования следуют из менделевских правил наследования. Правомерно будет сказать, что заключение при таком типе анализа сводится к тому, что в исследуемой родословной сегрегация того или иного заболевания не противоречит определенному менделевскому типу наследования.
Обычно в поле зрения медицинского генетика попадает семья, в которой есть больной или больные с предположительно наследственным заболеванием. Для такой семьи обычно собирается родословная, т.е. устанавливаются предки родителей и их родственники и описывается статус их здоровья, а также другие сведения. Принято представлять родословную графически, используя для этого определенные символы, изображенные на рис. 5-1.

Рис. 5-1. Некоторые символы, принятые при составлении родословных
Именно такая родословная становится предметом анализа медицинского генетика.
В 1966 г. появилось первое издание книги В. МакКьюсика «Менделевское наследование у человека. Каталог аутосомно-доминантных, аутосомно-рецессивных и Х-сцепленных фенотипов», в котором были собраны сведения о различных, нормальных и патологических, состояниях у человека, которые обнаруживают менделевское наследование. В абсолютном большинстве случаев установление типа наследования заболеваний, представленных в этом каталоге, основывалось на указанных выше принципах. В связи с нестрогостью этих принципов некоторые заболевания переходили из одного каталога в другой по мере накопления материала и появления новых изданий. В настоящее время в этой книге, которая перестала переиздаваться с 1994 г. и существует в постоянно пополняющейся версии в Интернете (адрес сайта: http://www.ncbi.nlm.nih.gov/Omim/), представлены описание и библиография более 20 тыс. фенотипов, из них с установленным типом наследования считается около 5 тыс. Основную часть описанных фенотипов составляют те, что наследуются аутосомно-доминантно и аутосомно-рецессивно, их более 4,5 тыс. Стоит добавить, что заболевания с этими типами наследования составляют в соответствующих каталогах примерно половину списков. За 44 года, прошедших с момента первого издания книги МакКьюсика, число описаний менделирующих фенотипов у человека выросло более чем в 10 раз.
АУТОСОМНО-ДОМИНАНТНОЕ НАСЛЕДОВАНИЕ
В настоящее время известно несколько тысяч аутосомно-доминантно наследующихся фенотипов. Мы остановимся на некоторых общих принципах их выявления с помощью генетического подхода.
На рис. 5-2 представлена родословная, в которой наследуется синдром Марфана - аутосомно-доминантное заболевание, ген которого FBN1 (ген фибриллина) картирован в хромосоме 15. Синдром Марфана проявляется системным поражением соединительной ткани, так как дефектный белок входит в состав межклеточного вещества соединительной ткани. Больные с синдромом Марфана обычно высокого роста, с длинными конечностями и арахнодактилией пальцев. Кроме того, у больных наблюдаются кифосколиоз, миопия высокой степени или подвывих хрусталика, расширение корня аорты.
В родословной три поколения. В первом поколении был болен отец (I1), но к моменту исследования семьи он умер. Среди семи его детей двое больных (дочь - II2 и сын - II5) и пятеро здоровых. II2 состоит в браке со здоровым мужчиной, у них семеро детей, из которых больны четверо (III2, III4, III5, III6) - три девочки и один мальчик, а трое здоровы. Здоровый сын первого больного (II7) состоит в браке со здоровой женщиной (II8). У них восемь детей, все они здоровы.
Приведенная выше родословная демонстрирует требования, предъявляемые к родословным с аутосомно-доминантным наследованием. Во-первых, один из родителей больных детей также должен быть болен. Во-вторых, болезнь должна встречаться у людей обоего пола. В-третьих, согласно менделевским правилам наследования, половина детей больного родителя должна быть больна, и риск, который составляет 50%, остается постоянным для каждого последующего ребенка. Однако данное требование не жесткое, поскольку при небольшом числе детей в семье могут случайным образом наблюдаться заметные отклонения от ожидаемого отношения больных детей к здоровым. В-четвертых, должна наблюдаться передача заболевания от отца к сыну, что исключает сцепленное с полом наследование. В-пятых, у здоровых потомков больного все дети должны быть здоровы.
В действительности даже в тех случаях, когда сталкиваются с аутосомнодоминантным заболеванием в семье, далеко не всегда удается наблюдать родословную, подобную той, что приведена на рис. 5-2. Для этого есть несколько причин.

Рис. 5-2. Родословная, в которой аутосомно-доминантно наследуется синдром Марфана
Одна из основных - это вновь возникающие мутации в отдельных половых клетках одного из родителей (при выполнении программы «Геном человека» установлено, что в абсолютном большинстве случаев новые мутации возникают в гаметогенезе у мужчин). При таком наследовании больной с аутосомно-доминантной патологией будет единственным случаем заболевания в семье. Очевидно, что для такого больного шанс передать мутацию своим детям будет обычным для аутосомно-доминантного типа наследования, т.е. равен 50%.
Вторая причина отклонения от правил аутосомно-доминантного наследования - мозаицизм зародышевых клеток (мозаицизм означает присутствие двух или более генетически различных линий клеток в одном организме). Такой мозаицизм возникает на ранних стадиях развития организма в результате появления мутации в одной из клеток зародышевого пути в момент его обособления. Поскольку клетки зародышевого пути клонируются, мутация может оказаться в большей или меньшей части зрелых половых клеток, и как результат возможно появление в семье здоровых родителей детей с аутосомно-доминантными заболеваниями. Именно поэтому соматическим мозаицизмом объясняются повторные случаи ахондроплазии, несовершенного остеогенеза и других аутосомно-доминантных заболеваний у детей, родители которых клинически совершенно здоровы.
АУТОСОМНО-РЕЦЕССИВНОЕ НАСЛЕДОВАНИЕ
По данным «Каталога наследственных признаков человека» (OMIM), известно несколько тысяч фенотипов с аутосомно-рецессивным типом наследования. Примерно 50% этих фенотипов представлено болезнями, наследующимися аутосомно-рецессивно. Как и аутосомно-доминантные, аутосомно-рецессивные заболевания поражают все органы и системы человека и, следовательно, чрезвычайно разнообразны по своим проявлениям.
В силу редкости аутосомно-рецессивных заболеваний, а также тяжести течения многих из них в абсолютном большинстве случаев родители больных детей являются гетерозиготными носителями и клинически здоровы. То, как сегрегирует аутосомно-рецессивный признак, можно увидеть в табл. 4-1. 1/4 детей в браке гетерозиготных родителей должны быть гомозиготами по нормальному доминантному аллелю, 1/2 - гетерозиготами и 1/4 - гомозиготами по мутантному аллелю. Риск появления больного с аутосомно-рецессивным заболеванием в семье, где родители являются гетерозиготными носителями мутантного гена, составляет 25% и не меняется для любой беременности этой супружеской пары. Родословные больных с аутосомно-рецессивным заболеванием обычно невыразительны: родители и все ближайшие родственники больного здоровы, могут быть больны братья и сестры (оба пола поражаются одинаково часто). Если больной с рецессивным заболеванием вступает в брак, то его партнер в большинстве случаев является нормальной гомозиготой, и поэтому все дети в таком браке здоровы, но являются гетерозиготными носителями. Нередко в родословных с аутосомно-рецессивными заболеваниями родители больных оказываются близкими родственниками (рис. 5-3).
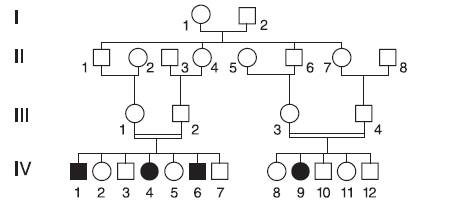
Рис. 5-3. Аутосомно-рецессивное наследование
На рис. 5-3 представлена родословная, в которой наследуется редкое аутосомнорецессивное заболевание - синдром Коккейна (OMIM 216400). Характерными признаками синдрома Коккейна являются карликовость, раннее старение, пигментная дегенерация сетчатки, атрофия зрительных нервов, тугоухость, умственная отсталость, повышенная чувствительность к свету и др. Ген синдрома картирован на хромосоме 5. Его белок участвует в репарации тиминовых димеров ДНК, возникающих после воздействия ультрафиолета.
Родители больных детей, как III1 и III2, так и III3 и III4, являются двоюродными сибсами. Кроме того, обе семьи с больными детьми родственны друг другу, так как II1, II4, II5 и II8 являются сибсами. Объяснение большей частоты близкородственных браков в семьях, где есть больные с аутосомно-рецессивным заболеванием, очень простое. Чем реже рецессивный ген встречается в популяции, тем меньше шансов, что оба родителя будут гетерозиготами по этому гену, так как вероятность такой супружеской пары равна произведению вероятностей возможности быть гетерозиготным носителем для каждого партнера. Так, при частоте гетерозиготного носительства гена фенилкетонурии, равной примерно 0,02, - вероятность брака гетерозиготных носителей составит 0,0004. Иными словами, одна из 2500 супружеских пар представлена гетерозиготными носителями (так как риск рождения больного ребенка для такой пары составляет 1/4, частота фенилкетонурии в популяции как раз и составляет 1:10 000). В том случае, когда один из супругов является гетерозиготным носителем гена фенилкетонурии (вероятность 0,02), а второй супруг является ему родственником, вероятность для второго супруга быть гетерозиготным носителем того же гена зависит от степени родства супругов. Для двоюродных сибсов она составляет, например, 25%, что в 12,5 раза выше, чем если бы супруги не были родственниками. Из приведенных расчетов также следует, что чем реже встречается рецессивный ген в популяции, тем большую долю среди семей с больными детьми будут составлять семьи, родители в которых являются близкими родственниками.
Итак, для аутосомно-рецессивного наследуемого признака характерно: носитель аутосомно-рецессивного признака является гомозиготой по мутантному аллелю соответствующего гена (если мутантные аллели у такого носителя являются разными, то его называют компаундной гетерозиготой), родители индивидуума с аутосомно-рецессивно наследуемым признаком (заболеванием) этого признака не имеют, но являются носителями мутантного гена. Признак могут проявить только братья и сестры (сибсы) этого индивидуума. Доля сибсов с рецессивным признаком в такой семье составляет 25%. Рецессивно наследуемый признак проявляется одинаково у сибсов разного пола. Для редких аутосомно-рецессивных заболеваний характерно, что родители больных детей значительно чаще, чем если бы это происходило случайно, являются близкими родственниками.
НАСЛЕДОВАНИЕ, СЦЕПЛЕННОЕ С Х-ХРОМОСОМОЙ
Гены, локализованные в Х-хромосоме, как и признаки, которые они контролируют, называются Х-сцепленными. Число известных Х-сцепленных заболеваний меньше, чем аутосомных, гены которых разбросаны по 22 аутосомам. Тем не менее известно около 300 генов, локализованных в Х-хромосоме, мутации в которых вызывают наследственные болезни. Всего же в Х-хромосоме выявлено более 600 генов, и, вероятно, это число не окончательное.
Прежде чем перейти к изложению наследования, сцепленного с Х-хромосомой, и особенностям родословных при этих типах наследования, следует напомнить, что у человека пол гетерогаметен. Женщины имеют во всех клетках две Х-хромосомы, а мужчины - одну Х- и одну Y-хромосому. Y-хромосома не гомологична Х-хромосоме, в ней содержится небольшое число генов. Мужчины являются гемизиготами по Х-хромосоме и большинству содержащихся в ней генов. Наследование половых хромосом происходит так же, как и простых менделевских признаков, схема которого представлена в виде решетки Пеннета (табл. 5-5).
Таблица 5-5. Схема наследования пола

Из табл. 5-5 видно, что при случайном объединении гамет должно образоваться равное количество зигот женского и мужского пола.
Поведение генов, которые расположены в половых хромосомах, строго соответствует поведению самих хромосом. Если мутантный ген находится в одной из Х-хромосом матери (например, в хромосоме Х1), то эту Х-хромосому получат половина сыновей и половина дочерей матери-носительницы. Если ген отвечает за рецессивное заболевание, то сама мать должна быть здорова, так как у нее есть вторая нормальная Х-хромосома, но оно разовьется у той половины сыновей, которые получили хромосому Х1 с мутантным геном, поскольку Y-хромосома не гомологична Х-хромосоме, и в ней просто не содержится пар абсолютному большинству генов Х-хромосомы. У дочерей, получивших от матери измененную хромосому, заболевание также не разовьется, так как они получат нормальную вторую Х-хромосому от отца. Таким образом, если мать является носительницей сцепленного с Х-хромосомой рецессивного гена, риск быть больным у ее сыновей составит 1/2, или 50%, а у ее дочерей - 0%. В то же время риск быть гетерозиготными носительницами у дочерей равен 50%.
Для рецессивных Х-сцепленных заболеваний характерно, что наследуют их мужчины - родственники друг другу по материнской линии. Это могут быть двоюродные братья, матери которых родные сестры, или дяди и племянники больного по материнской линии и т.д. (рис. 5-4).

Рис. 5-4. Х-сцепленное рецессивное наследование
В том случае, если отец болен, то все его сыновья будут здоровы, а все дочери - облигатными гетерозиготными носительницами Х-сцепленного гена. Такая ситуация бывает Рис. 5-4. Х-сцепленное рецессивное наследование тогда, когда Х-сцепленное рецессивное заболевание не очень резко снижает приспособленность больного. Примером подобного заболевания является Х-сцепленный рецессивный ихтиоз, или цветовая слепота к красному цвету, наследующаяся Х-сцепленно.
Любые Х-сцепленные рецессивные заболевания значительно чаще поражают мужчин, женщины болеют крайне редко, так как чтобы заболела женщина, должен болеть ее отец, а мать должна быть гетерозиготой по мутантному гену.
Для Х-сцепленных рецессивных, как и для аутосомно-доминантных заболеваний, свойственно возникновение части случаев в результате вновь возникших мутаций. Однако в отличие от аутосомно-доминантных заболеваний, когда при летальности этого заболевания на ранних этапах онтогенеза все его случаи объясняются вновь возникшими мутациями, при Х-сцепленных рецессивных заболеваниях такого не бывает, так как у женщин - гетерозиготных носительниц летального Х-сцепленного рецессивного гена - этот ген не проявляется, поскольку имеется его нормальная копия во второй Х-хромосоме. Еще в 1935 г. Холдейн показал, что при летальности Х-сцепленного рецессивного заболевания 1/3 больных появляется в результате вновь возникших мутаций, при условии одинаковой частоты мутирования в женских и мужских половых клетках. В настоящее время показано, что большая часть точковых мутаций в Х-хромосоме возникает во время сперматогенеза. В этом случае доля вновь возникших мутаций, обусловливающих Х-сцепленное рецессивное летальное заболевание (например, миопатию Дюшенна), будет меньше. Данная проблема чрезвычайно важна для медико-генетического консультирования, когда необходимо решить, является ли мать больного ребенка с Х-сцепленным рецессивным летальным заболеванием гетерозиготной носительницей мутантного гена или это случай вновь возникшей мутации. В первом варианте риск заболевания у ее следующего сына будет равен 50, а во втором - 0%.
Х-сцепленные рецессивные признаки обусловлены мутациями в Х-хромосоме. Такие мутации проявляются обычно только у мужчин, но не у женщин (за редкими исключениями). У 1/2 сыновей женщины, носительницы такой мутации, проявится рецессивный признак (заболевание), а 1/2 ее дочерей будут облигатными носительницами соответствующей мутации. Все дочери пораженного мужчины будут являться облигатными носительницами мутации.
Х-сцепленных доминантных признаков (в том числе и заболеваний) немного. Примером такого заболевания является D-резистентный гипофосфатемический рахит, при котором нарушена резорбция фосфатов в почечных канальцах и, как следствие, нарушена оссификация длинных трубчатых костей, в результате происходит их искривление, что особенно характерно для нижних конечностей. Еще одно Х-сцепленное доминантное заболевание - недержание пигмента (incontinentia pigmenti), проявляющееся нарушением пигментации кожи, а также глазными и неврологическими нарушениями и аномалиями зубов. Особенностью этого заболевания является то, что все гемизиготные мужчины, носители мутантного гена, поражаются настолько сильно, что погибают внутриутробно. В результате только женщины, имеющие это заболевание, попадают в поле зрения врачей, и возникает необходимость отличать Х-сцепленное доминантное наследование от аутосомно-доминантного наследования заболевания, ограниченного полом, такого, например, как рак молочной железы, обусловленный мутацией в гене BRCA1.
Если мутантный ген отвечает за Х-сцепленное доминантное заболевание, то будут больны 1/2 дочерей и 1/2 сыновей больной матери, т.е. как при аутосомнодоминантном заболевании, риск заболеть будет одинаковым для детей обоего пола - 1/2, или 50%. В этом случае родословная не позволяет различить два типа наследования. Заподозрить по родословной Х-сцепленное доминантное заболевание позволяет родословная, в которой болен отец. Поскольку отец передает свою Х-хромосому всем дочерям, то при достаточно большом числе больных девочек, а также большом числе здоровых мальчиков в семье предположение об Х-сцепленном доминантном заболевании, наследующемся в этой семье, может быть обоснованным.
Y-СЦЕПЛЕННОЕ НАСЛЕДОВАНИЕ
В Y-хромосоме найдено небольшое число генов, основная масса которых вовлечена в детерминацию мужского пола, или имеет отношение к сперматогенезу. Наследование генов и соответственно признаков, сцепленных с Y-хромосомой, называется голандрическим. В этом случае ген (признак) от отца передается только всем сыновьям, но не дочерям.
При Y-сцепленном наследовании ген, локализованный в Y-хромосоме, передается только сыновьям, но не дочерям, т.е. наследование признака происходит исключительно по мужской линии.
ПСЕВДОАУТОСОМНОЕ НАСЛЕДОВАНИЕ
Этот тип наследования называют еще частично сцепленным с полом. Так наследуются гены, которые расположены в псевдоаутосомных областях Х- и Y-хромо сом, т.е. на самых концах коротких плеч этих хромосом. Псевдоаутосомные области Х- и Y-хромосом гомологичны и спариваются в мейозе I. Гены, которые находятся в этих псевдоаутосомных областях, могут за счет кроссинговера переноситься из одной хромосомы в другую. В результате признаки, которые контролируют эти гены, в части родословной могут выглядеть как Y-сцепленные, а в другой - как Х-сцепленные. Так наследуется дисхондростеоз, или синдром Лери-Вейля, обусловленный мутациями в гене SHOX, который локализован в псевдоаутосомной области.
Идентификация генов, ответственных за менделирующие признаки
Еще 50 лет назад установление типа наследования для менделирующих признаков было практически конечным результатом исследования. После этого нормальные менделирующие, а значит, полиморфные признаки использовались в популяционной генетике человека для изучения генетической структуры популяций, а тип наследования для патологических признаков, или менделирующих заболеваний, служил основанием для расчета повторного риска в медико-генетических консультациях для семей с соответствующими заболеваниями. Употребленный термин «полиморфизм» обозначает, что в популяции ген может существовать в разных аллельных вариантах, а частота самого редкого варианта превышает 1%. В настоящее время понятие «полиморфизм» может относиться не только к разным аллелям гена, но и к другим последовательностям ДНК, в том числе некодирующим, если они встречаются в популяции в разной форме.
В международной программе «Геном человека» доминировали три основные задачи:
Создание генетической карты предполагало установление последовательности расположения генетических маркеров (в этом качестве использовались различные, преимущественно ДНК-полиморфизмы, т.е. наследуемые вариации в структуре ДНК) по длине всех хромосом с определенной плотностью - на достаточно близком расстоянии друг от друга. Такая генетическая карта должна была облегчить картирование всех генов человека, особенно генов наследственных болезней, что являлось одной из основных целей указанной программы. За короткое время было картировано несколько тысяч генов (рис. 5-5, см. цв. вклейку).
В качестве примера на рис. 5-5 представлена упрощенная модифицированная карта хромосомы 1, созданная специальной программой NCBI Map Viewer (www.ncbi.nlm.nih.gov/mapview/map_search), из которой удалены некоторые столбцы. Она состоит из ряда частей, в том числе из цитогенетической карты хромосомы 1, контигов для этой хромосомы (что такое контиги - будет сказано ниже), кластеров экспрессирующихся последовательностей (EST), секвенированных участков, символов картированных генов и их названий. Показано только небольшое число генов из тех, что картированы на хромосоме 1.
В программе «Геном человека» успешно были решены и две другие задачи - создание физической карты генома и его сиквенс. Остались неотсеквенированными примерно 300 участков генома относительно небольшого размера, для которых существующие методы сиквенса неэффективны.
ГЕНЕТИЧЕСКИЙ ПОЛИМОРФИЗМ
Установление локализации гена наследственной болезни, как, впрочем, и любого другого гена, возможно в том случае, когда удается установить или предположить фазу сцепления для исследуемого гена и полиморфного маркерного локуса. Это возможно при условии, что носитель гена наследственной болезни будет гетерозиготен по аллелям маркерного локуса. Таким образом, для анализа сцепления пригодны только те маркерные локусы, которые обнаруживают выраженный полиморфизм. Первый генетический полиморфный признак у человека был обнаружен Ландштейнером в 1900 г. Это была система группы крови АВО. До 1955 г. у человека было известно только несколько полиморфных генетических систем - преимущественно разные группы крови. В 1955 г. Смитис описал метод электрофореза в крахмальном геле, который позволял разделять белки по их заряду и молекулярной массе. Благодаря использованию этого метода Смитису удалось показать, что полиморфным является также сывороточный белок гаптоглобин. Было установлено, что электрофоретические варианты гаптоглобина наследуются как кодоминантные признаки. Вскоре генетический полиморфизм был обнаружен и для некоторых других сывороточных белков, а дополнение электрофореза методами определения ферментативной активности позволило установить, что полиморфизм свойствен также многим эритроцитарным, лейкоцитарным ферментам и ферментам плазмы крови. К 70-м годам было известно не менее 100 белковых полиморфизмов, которые можно было выявить с помощью различных вариантов электрофореза. К сожалению, большая часть белковых полиморфизмов оказалась мало пригодной для анализа сцепления с генами наследственных болезней, но сыграла исключительную роль в изучении генетической структуры популяций человека. Иные возможности для изучения сцепления и картирования генов открыли ДНК-полиморфизмы.
Одним из первых был обнаружен полиморфизм длины рестрикционных фрагментов (ПДРФ). Каждая рестриктаза разрезает молекулу ДНК, распознавая специфическую для нее последовательность нуклеотидов, которая называется сайтом рестрикции. Если в сайте рестрикции, независимо от того, где он находится, возникает мутация и происходит изменение последовательности нуклеотидов, то рестриктаза не узнает такой измененный сайт и не разрезает в этом месте молекулу ДНК. В то же время рестриктаза продолжает разрезать соседние неизмененные сайты рестрикции. В результате в мутантной ДНК один из фрагментов рестрикции оказывается длиннее, чем соответствующие фрагменты в немутантной ДНК. Для того чтобы выявить наличие полиморфного по рестрикции сайта в ДНК, ее после обработки рестриктазой подвергают электрофорезу. Во время электрофореза фрагменты ДНК распределяются в соответствии со своей молекулярной массой.
Обычно после рестрикции геномной ДНК образуются сотни тысяч фрагментов. Эти фрагменты денатурируются, т.е. превращаются в однонитевые и переносятся на нитроцеллюлозный фильтр, с которым прочно связываются, эта процедура называется блоттингом по Саузерну. Поскольку некоторые рестриктазы разрезают ДНК достаточно часто, то поиск полиморфизма можно вести с помощью различных ДНК-зондов, предварительно меченных радиоактивным фосфором, или флюоресцентной метки. Это могут быть зонды к участкам генов или любым другим последовательностям ДНК, локализация которых известна. Зонды соединяются с комплементарными последовательностями ДНК. В тех случаях, когда определенный зонд выявляет у разных людей разное количество меченых фрагментов или фрагментов с разной молекулярной массой, можно предполагать наличие рестрикционного полиморфизма в определенном локусе ДНК. Таким образом, можно обнаруживать мутации в генах, если эти мутации меняют сайт рестрикции для определенных рестриктаз (рис. 5-6).
В настоящее время известны сотни рестриктаз, которые распознают разные последовательности нуклеотидов. В распоряжении исследователей имеются тысячи ДНК-зондов к разным участкам генома. Использование этих возможностей привело к выявлению многих тысяч полиморфных сайтов в человеческом геноме. В абсолютном большинстве случаев ПДРФ представляет собой двухаллельную систему (наличие или отсутствие сайта рестрикции), тем не менее его высокая частота была успешно использована при картировании многих генов наследственных болезней, в том числе муковисцидоза, нейрофиброматоза I типа и многих других.
Значительно большее количество аллелей и, следовательно, большее генетическое разнообразие, а также информационная ценность у еще одного типа ДНКполиморфизма, который называется «варьирующее число тандемных повторов», или VNTR полиморфизм. В геноме человека встречаются различные типы ДНК, в том числе так называемые минисателлитные повторы. Эти повторы разбросаны по геному, их основная последовательность может включать до 80 нуклеотидов, а число повторов основной последовательности может варьировать в широких пределах. Выявление VNTR полиморфизма начинается так же, как и выявление ПДРФ, с разрезания геномной ДНК той или иной рестриктазой, электрофореза фрагментов, их денатурации и блоттинга по Саузерну. Отличие заключается в том, что для гибридизации используются ДНК-зонды, специфичные для определенного минисателлитного повтора.

Рис. 5-6. ПДРФ, обусловленный полиморфным сайтом рестрикции. А1 и А2 гомологичные хромосомы. Рестриктаза разрезает в сайте b хромосому А1, но не А2. В результате на Саузерн-блоте с зондом к определенному участку хромосомы А легко различаются все три генотипа (Из I.D. Young. Medical Genetics. - Oxford Univ. Press, 2005. - P. 142, модифицировано)
Еще более информативным ДНК-полиморфизмом являются микросателлитные тандемные повторы. Так же как и минисателлитные, микросателлитные повторы отличаются по числу повторяющихся последовательностей. Однако в случае микросателлитов базовая последовательность состоит всего из двух, трех или четырех нуклеотидов. Кроме того, они более часто встречаются в геноме и распределены по нему более равномерно. Отличается также способ их детекции. Микросателлитные повторы обычно выявляются с помощью ПЦР. Для этого необходимо знать последовательность нуклеотидов, предшествующих и следующих за повтором, чтобы выбрать подходящие праймеры, однако секвенирование генома так далеко продвинулось вперед, что найти такую последовательность не представляет особого труда ни для одного региона генома. Это очень удобный полиморфизм для изучения сцепления не только потому, что полиморфных сайтов в геноме очень много, но еще и потому, что во многих случаях полиморфизм проявляется большим количеством аллелей, и гетерозиготность при этом типе полиморфизма очень высокая.
В настоящее время часто для анализа сцепления используется так называемый однонуклеотидный полиморфизм (ОНП). ОНП могут представлять собой замещения, делеции или инсерции одного нуклеотида в последовательность нуклеотидов ДНК (рис. 5-7, см. цв. вклейку).
ОНП теоретически наблюдается с частотой 1 на 100-300 нуклеотидов и по форме представляют собой диаллельный полиморфизм. При такой частоте ОНП соответствуют всем требованиям, которые предъявляются к маркерам для широкомасштабного исследования сцепления. Кроме того, ОНП должен встречаться несколько раз даже в небольших по размеру генах. В этом случае ОНП может быть собственно мутацией, обусловливающей наследственное заболевание. Изучение распространенности ОНП в геноме человека шло под эгидой специально созданного консорциума. В настоящее время в геноме человека идентифицировано несколько миллионов ОНП. Вскоре после начала работы по выявлению ОНП стало ясно, что многие ОНП, расположенные в одной хромосоме, тесно сцеплены, образуя определенные блоки или гаплотипы. Для изучения этого феномена был организован специальный международный проект под названием HapMap. Стало ясно, что для идентификации такого блока тесно сцепленных ОНП, достаточно определить лишь несколько ОНП (более полную информацию о проекте можно получить на сайте http://snp.cshl.org). В настоящее время существуют коммерческие чипы, содержащие 500 тыс. и более картированных ОНП, которые используются для изучения сцепления генов с малым эффектом.
Для успешного картирования генов наследственных болезней выявления многочисленных и удобных для картирования ДНК-полиморфизмов различных типов было недостаточно. Эти маркеры надо было локализовать на генетической карте, установив их взаимное расположение, что стало возможным благодаря тому, что картирование ДНК-полиморфных маркеров было проведено большинством исследователей на одной и той же выборке семей. Эта выборка включает 61 многопоколенную семью, с большим числом членов в каждой семье, отобранных в исторических провинциях Франции Центром по изучению полиморфизма у человека (Centre d?Etude du Polymorhisme Humain - CEPH). Все исследователи, которые занимались картированием ДНК-полиморфизмов, могли получить ДНК от членов этих семей. Как только первые ДНК-маркеры были картированы по всем хромосомам, работа по картированию новых маркеров ускорилась, так как их надо было просто встроить в уже имевшуюся карту для каждой хромосомы. В очень короткий срок удалось создать генетическую карту для всего генома, в которой расстояние между ближайшими ДНК-маркерами не превышало в среднем 5 сантиморган (сМ). Были созданы коммерческие наборы для определения 300 и более ДНК-полиморфизмов в геноме, которые позволяли провести так называемый геномный скрининг и относительно легко картировать гены наследственных болезней. Кроме того, столь плотно заполненная маркерами генетическая карта позволила уменьшить количественные требования к семейному материалу, пригодному для изучения сцепления генов.
Параллельно с исследованиями по программе «Геном человека», имеющей глобальное значение, шло генетическое картирование локусов, ответственных за наследственные болезни. Эта работа значительно облегчалась благодаря созданию генетической карты ДНК-маркеров в рамках программы. Картирование генов наследственных болезней является принципиально важным для медицины, так как позволяет проводить непрямую диагностику соответствующих наследственных болезней. Кроме того, генетическое картирование является необходимым шагом в идентификации и последующем клонировании того или иного гена наследственной болезни, изучения его структуры, природы мутаций в этом гене и в перспективе открывает возможности манипуляций с этим геном, например в генотерапевтических целях.
Разные хромосомы в мейотических делениях ведут себя независимо друг от друга, что составляет хромосомную основу для выполнения менделевского правила независимого наследования признаков. В то же время гены, расположенные в одной хромосоме, наследуются преимущественно как одна группа, т.е. сцепленно, кроме тех случаев, когда происходит кроссинговер. В результате кроссинговера происходит обмен одинаковыми участками гомологичных хромосом, и в рекомбинантной хромосоме создается новая комбинация генов, часть из которых происходит из одной, а другая часть - из другой гомологичной хромосомы каждого из родителей. Следует помнить, что в среднем на хромосому приходится от одной до трех рекомбинаций. Рекомбинации происходят во время оогенеза и сперматогенеза (в мейозе I), так что ребенок получает рекомбинантные хромосомы как от матери, так и от отца.
Установить, есть ли сцепление между геном, вызывающим аутосомнодоминантное заболевание, и каким-либо генетическим полиморфным маркером, можно на примере семьи, в трех поколениях которой наследуется вульгарный ихтиоз. При обследовании членов этой семьи для выяснения генотипов каждого по выбранному генетическому маркеру получена родословная с описанием генотипов каждого члена семьи (рис. 5-8).
Судить о том, сцеплен ли избранный маркерный локус с геном ихтиоза, можно только по третьему поколению, но для этого необходимо сделать предположение, с каким аллелем маркерного локуса наследуется вместе ген ихтиоза у II2, т.е. у больного отца. В трехпоколенной родословной это сделать просто. Как видно на рис. 5-8, ген ихтиоза наследуется вместе, т.е. находится в одной хромосоме, с аллелем 1 маркерного локуса (такое состояние называется фазой притяжения), с аллелем 2 маркерного локуса ген ихтиоза находится в фазе отталкивания. Если теперь проанализировать генотипы всех детей из поколения III, то становится ясно, что все дети, кроме III6, являются не рекомбинантами, а III6, напротив, рекомбинанта. В этом случае частота рекомбинаций (обозначается как θ) составляет 1 из 7, т.е. 0,14.
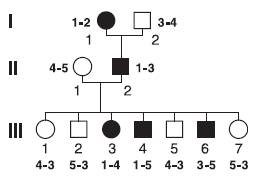
Рис. 5-8. Семья, в которой сегрегирует аутосомно-доминантный ихтиоз (пораженные обозначены черными кружочками и квадратами) и ДНК полиморфный маркер (генотипы каждого члена семьи указаны жирным шрифтом)
Анализ частоты рекомбинаций становится значительно сложнее в случаях с рецессивными заболеваниями или с двухпоколенными семьями. Тогда для установления сцепления используется метод, основанный на максимально правдоподобной оценке частоты рекомбинаций (θ). Такая оценка основывается на относительной вероятности наличия сцепления между изучаемыми локусами в каждой конкретной семье (PR). PR рассчитывается как отношение вероятностей сцепления изучаемых локусов в данной семье, с частотой рекомбинаций между этими локусами (θ), варьирующей от 0 до 0,5, к вероятности, что в этой семье нет сцепления по изучаемым локусам и, следовательно, θ=0,5. Для удобства PR часто выражается через логарифм. Log10 этой относительной вероятности называется логарифмом шансов (log of odds) или значением логарифма шансов (lod score). Максимально правдоподобная оценка частоты рекомбинации θ между двумя изучаемыми локусами может быть получена путем нанесения на график сумм логарифма шансов для всех семей, на которых проводилось исследование, при разных значениях θ от 0 до 0,5. Значение θ, соответствующее пику кривой на графике, будет максимально правдоподобным. Считается, что сцепление между локусами установлено, когда сумма логарифмов шансов для исследованных семей окажется равной 3 или более. Логарифм шансов, равный 3, означает, что сцепление в 1 тыс. раз более вероятно, чем его отсутствие. Напротив, при значении логарифма шансов, равном -2, сцепление между локусами в 100 раз менее вероятно, чем его отсутствие, и может быть отвергнуто.
Если сцепление между локусами установлено, то частота рекомбинаций может быть легко превращена в расстояние на генетической карте, которое разделяет исследованные локусы. Считается, что 1% рекомбинаций соответствует расстоянию в 1 сМ. Следует заметить, что такое соответствие выполняется, когда локусы достаточно тесно сцеплены и частота рекомбинаций невысока. В противном случае между локусами могут возникать двойные или даже тройные кроссинговеры, и вычисленная частота рекомбинаций фактически оказывается заниженной. Упрощая ситуацию, можно от генетических расстояний перейти к физическим расстояниям между сцепленными генами, так, 1 сМ генетической карты примерно соответствует 1 млн пар нуклеотидов (п.н.). Однако такие переходы надо делать с большой осторожностью, так как известно, что частота рекомбинаций зависит от многих факторов. В частности, она ниже у мужчин по сравнению с женщинами, частота рекомбинаций также разная для разных районов каждой хромосомы, являясь максимальной в теломерных районах. Кроме того, установлено, что по длине хромосом выявляются так называемые горячие точки рекомбинаций.
В тех случаях, когда устанавливается сцепление между двумя локусами, невозможно ответить на вопрос, каково взаимное расположение локусов. Ответ на этот вопрос может быть получен в эксперименте с мультилокусным сцеплением. Классический вариант такого анализа, когда изучалось сцепление между тремя локусами, был разработан еще в 20-е годы прошлого столетия Морганом и его сотрудниками. Суть этого эксперимента очень простая. Допустим, требуется установить взаимное расположение локусов А, В и С. Можно получить следующие результаты:
-
расстояние между локусами А и С равно 10 сМ, расстояние между локусами А и В равно 4 сМ, расстояние между локусами В и С равно 6 сМ - локус В расположен между локусами А и С;
-
расстояние между локусами А и С равно 4 сМ, расстояние между локусами А и В равно 10 сМ, расстояние между локусами В и С равно 6 сМ - локус С расположен между локусами А и В;
-
расстояние между локусами В и С равно 10 сМ, расстояние между локусами А и В равно 4 сМ, расстояние между локусами А и С равно 6 сМ - локус А расположен между локусами В и С.
Если в распоряжении имеется достаточно большое количество локусов, расположенных в одной хромосоме, то, создавая последовательно комбинации по трем или большему числу локусов, можно построить генетическую карту целой хромосомы. Некоторым осложнением при выполнении такой процедуры создания генетической карты являются двойные кроссинговеры, которые происходят тем чаще, чем дальше расположены друг от друга изучаемые локусы. Если, однако, мы можем различать генотипически двойные кроссинговеры, то легко внести соответствующую поправку при определении генетического расстояния между изучаемыми генетическими локусами.
Для анализа сцепления гена наследственного заболевания, когда используются тысячи и даже сотни тысяч полиморфных генетических маркеров, плотно перекрывающих весь геном человека, широко используются различные компьютерные программы, существенно облегчающие эту работу. Наиболее известные из них LINKAGE и LIPED (эти и многие другие программы по анализу сцепления можно найти на сайте http://linkage.rockefeller.edu/soft/list2.html).
Многочисленные ДНК-полиморфизмы, обладающие высокой информативной ценностью, позволили использовать в ряде случаев для картирования генов наследственных болезней такой феномен, как неравновесность по сцеплению. Обычно генетические маркеры, обнаруживающие сцепление с генами наследственных болезней, встречаются в семьях с такой же частотой, как и в популяции, и инструментом для анализа сцепления является их сегрегация, зависимая или независимая от гена наследственного заболевания в конкретной семье. Однако в некоторых случаях, когда мутация в гене наследственного заболевания возникла относительно недавно и когда определенный аллель полиморфного генетического маркера очень тесно сцеплен с геном наследственного заболевания и находится с ним в фазе притяжения, это будет проявляться в том, что ген наследственного заболевания и какой-то аллель полиморфного генетического маркера будут встречаться в популяции всегда, или почти всегда, вместе. Неравновесие по сцеплению между разными ДНК маркерами и генами наследственных болезней найдено при муковисцидозе, хорее Гентингтона, фенилкетонурии, миотонической дистрофии и ряде других наследственных заболеваний.
МЕТОДЫ ФИЗИЧЕСКОГО КАРТИРОВАНИЯ ГЕНОВ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
Кроме генетического картирования, для локализации генов наследственных болезней человека используются разнообразные методы физического картирования генов. Самый простой подход физического картирования реализуется, когда тот или иной локус или ген заболевания оказывается ассоциированным с хромосомной аномалией - делецией, транслокацией или другой хромосомной аномалией, которая выявляется цитогенетически.
Картирование генов с помощью хромосомных мутаций
Если у больных с тем или иным наследственным заболеванием выявляются делеции примерно в одном и том же месте одной и той же хромосомы, это почти наверняка указывает, что ген наследственной болезни локализован в том районе хромосомы, который отсутствует во всех делециях, независимо от их размера. Таким образом было картировано, по крайней мере, несколько генов наследственных болезней, в том числе генов ретинобластомы, синдрома Ангельмана и др. Использование делеций для локализации генов было названо методом делеционного картирования. Сходным образом для установления локализации гена наследственной болезни могут быть использованы сбалансированные транслокации, когда у носителя такой транслокации диагностируется наследственное заболевание. Это позволяет предполагать, что один из разрывов хромосомы, предшествующий возникновению транслокации, нарушил структуру гена, локализованного в месте разрыва. Самым ярким примером, когда с помощью транслокации был картирован ген, является миопатия Дюшенна. Ген миопатии Дюшенна локализован в Х-хромосоме и обычно проявляется тяжелой миопатией у мальчиков. Было обнаружено несколько случаев типичной клинической картины миопатии у женщин. Они оказались связанными с транслокациями между Х-хромосомой и аутосомами, причем в Х-хромосоме разрыв всегда локализовался в районе хромосомы Хp21.2-p21.1. Это позволило резко сузить район поиска гена миопатии Дюшенна. Здесь необходимо подчеркнуть, что, кроме транслокации, у пораженных женщин должна была также наблюдаться преимущественная инактивация нормальной Х-хромосомы.
Картирование гена иногда может быть достигнуто за счет использования эффекта дозы гена. В случае делеции следует ожидать уменьшения продукта гена (это может быть прежде всего фермент) на 50%. Именно таким способом был картирован ген кислой фосфатазы эритроцитов в хромосоме 2. При дупликации, наоборот, можно ожидать увеличения на 50% активности ферментов, гены которых вовлечены в дупликацию. Самым известным примером картирования гена с помощью дупликации является супероксиддисмутаза, которая была картирована на хромосоме 21, так как ее уровень был постоянно повышен у больных с болезнью Дауна.
Картирование с помощью гибридизации in situ
К физическим методам картирования относится также метод гибридизации in situ. В том случае, когда известна последовательность ДНК интересующего локуса (теперь такая ситуация возникает нередко), эту последовательность можно использовать для гибридизации с хромосомами in situ, и место гибридизации будет однозначно указывать на локализацию локуса в определенном районе определенной же хромосомы. Обычно гибридизации in situ предшествует выделение мРНК из какого-либо органа или ткани для характеристики функциональной дифференциальной активности генов в соответствующем органе. На основе этих мРНК с помощью обратной транскриптазы получают кДНК. Такие кДНК представляют собой экзоны тех генов, которые обнаруживают активность в исследуемом органе. Эти кДНК можно использовать как основу для создания ДНК-зондов. Их можно метить флюресцентными красителями и применять для флюоресцентной гибридизации с препаратами прометафазных хромосом, которые обработаны соответствующим образом, для того чтобы добиться денатурации нитей ДНК (FISH-анализ). В этом случае ДНК-зонд гибридизуется с комплементарной последовательностью на хромосомах и по флюоресцентному свечению выявляется и локализуется ген, с которого транскрибировалась мРНК, послужившая исходным звеном всего анализа. Такую кДНК можно использовать также для скрининга геномной библиотеки, созданной на основе тотальной клеточной ДНК. После гибридизации кДНК с определенным районом в такой библиотеке можно получить и исследовать полную структуру гена, включая его экзоны, интроны и промотор.
Гибридизация соматических клеток и картирование генов человека
Для физического картирования широко используются также методы гибридизации соматических клеток. Обычно для гибридизации с клетками человека используются соматические клетки грызунов, чаще всего мыши. Для того чтобы облегчить слияние клеток разных видов, в культуральную среду добавляют вирус Сендай, инактивированный ультрафиолетовым облучением, или полиэтиленгликоль. Для отбора слившихся исходных клеток человека и мыши, клетки выращивают на специальной селективной среде, которая позволяет размножаться только гибридным клеткам (для этого в опытах по слиянию использовали линии клеток мышей с недостаточностью по некоторым ферментам, чаще всего с недостаточностью по тимидинкиназе). В только что образовавшихся гибридных клетках ядра содержат оба набора хромосом человека и мыши. Однако последующее размножение гибридных клеток приводит к постепенной утрате хромосом человека у гибридов, так что в результате остается только одна или даже фрагмент одной из хромосом человека. Когда метод гибридизации соматических клеток был только создан, тестировалось присутствие в гибридных клетках, в которых осталась одна или несколько хромосом, ферментов и других белков человека, которые по своим характеристикам отличались от соответствующих белков мыши, или клетки мыши были дефицитны по каким-то ферментам. Обычно из гибридов соматических клеток создавались панели, в которых присутствовали хромосомы человека в минимальных количествах и в самых разных комбинациях. Именно поэтому для установления локализации гена соответствующего фермента или другого белка человека не было необходимости использовать только те гибриды соматических клеток, которые содержали лишь одну хромосому человека. Вместе с тем сначала можно было заниматься картированием только тех генов человека, которые проявлялись на клеточном уровне, и невозможно было работать с генами, имевшими сложное фенотипическое проявление, где первичный дефект оставался неизвестным. Методы молекулярной генетики позволили решить эту проблему, и сейчас с помощью ПЦР, блоттинга по Саузерну и иных молекулярно-генетических методов можно тестировать гибридные соматические клетки на наличие в оставшихся хромосомах человека любых генов, независимо от того, известен продукт этих генов или нет (рис. 5-9).
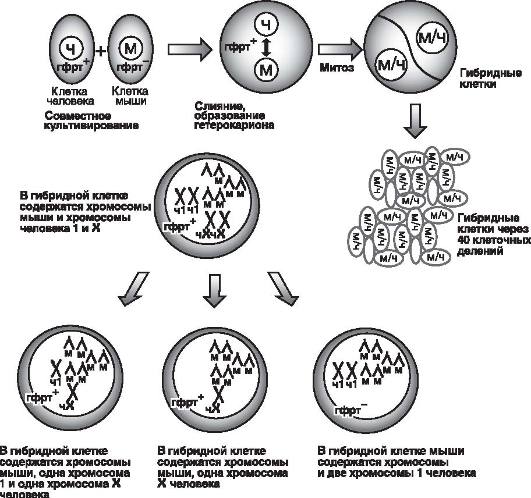
Рис. 5-9. Картирование генов человека с помощью гибридных клеток человек-мышь. Мышиные клетки, дефицитные по ГФРТ (гипоксантин-фосфорибозилтрансферазе), выращиваются в культуре in vitro вместе с нормальными клетками человека. С помощью вируса Сендай обеспечивается слияние мышиных и человеческих клеток. Образовавшиеся гибридные клетки через несколько десятков поколений теряют большую часть хромосом человека. Только в тех гибридных клетках, которые содержат Х-хромосому человека, выявляется активность ГФРТ. Из этого следует, что ген ГФРТ локализован в хромосоме Х человека
Недостатком метода картирования генов человека с помощью гибридных соматических клеток было то, что часто локализация генов устанавливалась с точностью до хромосомы. Разрешающие возможности в картировании генов таким путем были ограничены. Однако если в гибридных клетках осталась только одна хромосома человека, то, облучив такие гибридные клетки рентгеновскими или гамма-лучами, можно добиться фрагментации этой хромосомы и после получения новых гибридных клеток с помощью дополнительного слияния с клетками грызунов (после облучения гибридные клетки без дополнительного слияния с клетками грызунов погибают) уточнить локализацию интересующего исследователей гена, вплоть до небольшого фрагмента определенной хромосомы (картирование с помощью радиационных гибридов). Особенностью радиационных гибридов является то, что образующиеся после облучения фрагменты хромосомы человека не способны проходить митоз и теряются, если они не сливаются с хромосомами мыши. Эти фрагменты человеческих хромосом могут быть столь небольшими по размеру, что уже не обнаруживаются цитологически. Именно поэтому до проведения опытов по картированию необходимо специальными методами, в частности с использованием ДНК-полиморфных маркеров (обычно используется Aluполиморфизм, отсутствующий у мыши) уточнить, какие фрагменты определенной хромосомы человека сохранились в радиационных гибридах. Еще одной особенностью радиационных гибридов является то, что на них можно изучать сцепление между генами во фрагментах хромосом человека и даже оценивать генетическое расстояние между сцепленными локусами, исходя из того, насколько часто они присутствуют во фрагментах вместе или раздельно. При этом предполагается, что чем ближе расположены гены друг к другу на хромосоме, тем реже радиационноиндуцированные разрывы в хромосоме будут разделять сцепленные гены.
Идентификация генов человека с помощью позиционного клонирования и другими методами
Установление локализации гена наследственной болезни теперь может рассматриваться как подготовительный этап для его идентификации, т.е. выделения, клонирования, изучения структуры гена и последовательности нуклеотидов, а также обнаружения мутаций в этом гене, которые, собственно, можно рассматривать как этиологический фактор для соответствующего менделирующего наследственного заболевания. Классическая молекулярная генетика идентификацию гена обычно начинала с его первичных продуктов - мРНК или белка. Эти продукты позволяли получить фрагменты гена либо как кДНК, либо как последовательности нуклеотидов, соответствующие последовательности аминокислот белка (такая последовательность может быть короткой, и для ее создания вовсе не надо определять всю последовательность аминокислот в соответствующем белке, достаточно определить последовательность 1-2 десятков аминокислот). Эти фрагменты гена превращали в ДНК-зонды и с их помощью идентифицировали мутантный ген в геномных библиотеках или с помощью гибридизации in situ на препаратах хромосом. Такой путь идентификации гена, когда все начинается с мутантного белка, принято называть функциональной геномикой. Он был успешно применен для изучения генов, кодирующих фенилаланингидроксилазу, гемоглобин, VIII фактор свертывания крови, и некоторых других генов. Однако таких примеров можно привести немного, так как для большинства наследственных болезней их молекулярный патогенез остается неизвестным. Именно поэтому с середины 80-х годов ХХ века на смену функциональному клонированию пришли методы позиционного клонирования, при котором сначала картируется и изолируется ген заболевания, а затем по его структуре идентифицируется белок, который этот ген кодирует. Позиционное клонирование принципиально состоит из двух этапов: картирование гена насколько возможно более точное в определенном локусе хромосомы и идентификация внутри этого локуса гена-кандидата. Под локусом в этом случае понимают генетически картированный участок хромосомы или отрезок ДНК. Обычно стремятся, чтобы размер локуса, в котором находится ген-кандидат, не превышал 1 сМ, что соответствует примерно 1 млн п.н., в таком локусе может находиться около 10 генов. Позиционное клонирование предполагает выявление всех генов, находящихся в картированном локусе, с последующей идентификацией гена, ответственного за развитие заболевания. Для этого используются различные подходы. Первый из них - это конструкция контига. Контиг - набор перекрывающихся фрагментов клонированной ДНК, представляющих обычно небольшую часть генома. Для физического картирования контиг должен включать не только ген заболевания, но и фланкирующие его сцепленные генетические маркеры, которые были использованы для картирования локуса и выполняют роль реперных точек в процессе выявления генов в локусе. Сначала контиги конструировались из библиотек клонированной в векторах ДНК. Векторы - структуры, включающие ДНК для клонирования, которые, попадая в клетку хозяина, автономно размножаются, одновременно размножая клонируемую ДНК. Известно большое число разнообразных векторов, но чаще всего в этом качестве использовали искусственные хромосомы бактерий или дрожжей (BACs и YACs), которые могут инкорпорировать отрезки ДНК большого размера (вплоть до 1 млн п.н.), в отличие от таких векторов, как космиды, фаги или плазмиды, в которые можно встроить только относительно небольшие по размеру отрезки чужеродной ДНК. Теперь готовые YAC-контиги, подходящие для поиска гена заболевания в любой части генома, могут быть получены из базы данных генома человека. Такая ДНК, включенная в YAC, используется как зонд. При скринировании библиотек кДНК-зонд позволяет выявить все гибридизующиеся с ним геномные кДНК и проанализировать их после селективной амплификации и клонирования.
Еще один подход, использующийся для выявления генов в контиге, - это поиск CpG-островков, которые рассматриваются как практически обязательная часть 5?-конца многих генов и которые, вероятно, являются местами связывания факторов транскрипции. Эти кластеры обычно неметилированы и выявляются с помощью рестрикционных ферментов, чувствительных к метилированию, таких, например, как HpaII. Для выявления генов в контиге используются и другие методы, в том числе так называемый «захват экзонов», при котором гибридизующиеся с зондом участки ДНК вводятся в экспрессионную систему, где они могут образовывать РНК. После сплайсинга такой транскрипт РНК с помощью обратной транскриптазы превращается в кДНК, соответствующую с высокой вероятностью экзонам гена, содержащегося в контиге. Еще одним методом, позволяющим предположительно выявлять гены в контиге, является так называемый зооблотинг. В этом случае ДНК-зонд гибридизируется с ДНК, полученной от филогенетически разных видов. Предполагается, что гибридизация ДНК-зонда с большим числом ДНК разных видов является указанием на консервативность определенных фрагментов зонда, и, следовательно, они содержат кодирующие последовательности одного или нескольких генов.
После того как в контиге идентифицировано несколько генов, появляется возможность установить, какой из них является геном заболевания. Прямой подход предполагает секвенирование всех выявленных генов или генов, структура и функция которых может иметь отношение к проявлениям соответствующего моногенного заболевания. Секвенирование позволяет выявить тот ген, в котором наблюдается мутация, присутствующая у больных, но отсутствующая у их здоровых родственников, или в достаточно большой контрольной выборке. Если эта мутация вызывает сдвиг рамки считывания или делецию, то, скорее всего, она играет патогенетическую роль. В противном случае могут понадобиться исследования функциональной активности мутантного гена (рис. 5-10).

Рис. 5-10. Подходы, которые используются для позиционного клонирования (см. объяснения в тексте)
Секвенирование генома человека и идентификация в нем более 13 тыс. генов позволяют в некоторых случаях значительно упростить процедуру поиска генакандидата определенного менделирующего заболевания. Это делается с помощью специальных компьютерных программ анализа генома человека, из которых наиболее используемая - Ensemble (www.ensemble.org). Программа позволяет выявить все экспрессирующиеся последовательности и гены в интересующей исследователя области (локусе) генома, результаты сиквенса и структуру генов, а также их гомологию. Иногда этого оказывается достаточно, чтобы определить наиболее вероятный ген-кандидат.
В последнее время достигнуты большие успехи в разработке новых высокопродуктивных методов сиквенса генома человека. Для идентификации генов менделирующих заболеваний при небольшом числе больных создан метод сиквенса всего экзома в комбинации с методологией фильтрации результатов (поиск двух несинонимичных замен, мутаций сплайсинга и indel-вариантов, исключение полиморфизмов и т.д.). Вскоре идентификация генов редких менделирующих заболеваний может стать только технологической задачей, даже при наличии в семье лишь одного больного.
СПИСОК РЕКОМЕНДУЕМОЙ ЛИТЕРАТУРЫ
Гинтер Е.К. Медицинская генетика. - М.: Медицина, 2003. - 447 с.
Ньюссбаум Р.Л., Мак-Иннес Р.Р., Виллард Х.Ф. Медицинская генетика: Пер. с англ. - М.: ГЭОТАР-Медиа, 2010. - 608 с.
Young I.D. Medical Genetics. - Oxford: Oxford University Press, 2005. - 304 p.
Jorde L.B., Carey J.C., Bamshad M.J., White R.L. Medical Genetics. 2nd ed. - Mosby, 1999. - 372 p.