Наследственные болезни : национальное руководство / Под ред. Н. П. Бочкова, Е. К. Гинтера, В. П. Пузырева - Москва : ГЭОТАР-Медиа, 2012. - 936 с. (Серия "Национальные руководства") - ISBN 978-5-9704-2231-1 |
Аннотация
Национальные руководства - первая в России серия практических руководств по основным медицинским специальностям, включающих всю основную информацию, необходимую врачу для непрерывного последипломного образования.
Национальное руководство "Наследственные болезни" содержит актуальную, современную информацию о геноме человека, общих вопросах медицинской генетики, клинической генетике. Руководство состоит из двух частей, в которых излагаются теоретические и клинические вопросы медицинской генетики. В первой части представлены новейшие данные по теоретическим вопросам медицинской генетики. Сведения об организации и функциях генома, генов и хромосом изложены в понятной для врачей форме, но без излишнего упрощения. Во второй части представлены вопросы клинической генетики, а именно методы диагностики наследственных болезней (от клинического уровня до секвенирования ДНК и РНК), принципов лечения и профилактики отдельных нозологических форм. Поскольку в национальных руководствах по другим специальностям описаны многочисленные наследственные болезни, на них можно найти ссылки [см. "Перечень наследственных болезней (синдромов), описание которых представлено в других национальных руководствах" на компакт-диске]. Приложение к руководству на компакт-диске включает более полную информацию по некоторым главам, электронную версию руководства, обширный иллюстративный материал, приложения, перечень наследственных болезней, описанных в других руководствах, фармакологический справочник. В подготовке настоящего издания в качестве авторов-составителей и рецензентов принимали участие ведущие ученые разных специальностей: генетики, иммунологи, невропатологи, фармакологи, онкологи и другие специалисты. Все рекомендации прошли этап независимого рецензирования.
Руководство предназначено для врачей-генетиков, врачей лаборантов-генетиков, врачей смежных специальностей, интернов, ординаторов, аспирантов, особенно по таким дисциплинам, как педиатрия, акушерство-гинекология, нервные болезни.
Гриф
Национальное руководство рекомендовано Российским обществом медицинских генетиков и Ассоциацией медицинских обществ по качеству
Глава 15. Клиническая диагностика наследственных болезней
Общие замечания
Успешная диагностика наследственных болезней основана на знании и владении клинико-генеалогическим методом, использовании синдромологического подхода к диагностике наследственных болезней, результатах параклинических исследований, осведомленности об основных признаках, особенностях клинических симптомов наследственных нарушений и общих принципов клинической диагностики, особенностей осмотра и физикального обследования пациентов и их родственников. Важнейшая роль в диагностике наследственных болезней принадлежит лабораторным исследованиям: цитогенетическим, молекулярно-генетическим, биохимическим и др. (см. также гл. 16).
Термин «синдром» в клинической генетике употребляют для обозначения не только совокупности симптомов, объединенных единым патогенезом, но и болезней, составляющих самостоятельные нозологические единицы. Это обусловлено тем, что многие нозологические формы первоначально были описаны в качестве симптомокомплексов без понимания их этиологии. Хотя в дальнейшем была расшифрована наследственная этиология симптомокомплекса или синдрома, вплоть до его полной генетической характеристики (хромосомные болезни, генные болезни, митохондриальные болезни), за наследственными болезнями, первоначально описанными как синдромы, сохранился термин «синдром». Например, после расшифровки этиологии синдрома Клайнфелтера была попытка называть его болезнью Клайнфелтера, но она оказалась безуспешной. Именно поэтому термины «болезнь» и «синдром» для наследственных нарушений равнозначны. Для обозначения некоторых нозологических форм употребляют оба термина (например, болезнь Дауна, синдром Дауна). Люди, страдающие синдромом Дауна, и их опекуны по понятным причинам чувствительны к терминам, используемым для описания этого хромосомного нарушения. В связи с этим после идентификации хромосомной основы синдрома Дауна в 1959 г. постепенно стали применять термин «трисомия 21».
Особенности клинических признаков наследственных болезней
Любой вид патологических изменений (инфекционное заболевание, ожоги, травмы) сопровождается конкретной клинической картиной, в основе которой лежит взаимодействие повреждающего фактора с организмом. Знание этих закономерностей помогает врачу в диагностике заболеваний и лечении больных. Наследственные нарушения, несмотря на огромное нозологическое многообразие, имеют специфические черты, которые врач должен использовать в качестве ориентиров в диагностическом поиске.
В основе клинических признаков наследственных заболеваний лежат закономерности проявления генов и их взаимодействия.
Ниже изложены общие признаки наследственных нарушений, позволяющие врачу заподозрить роль наследственных факторов в этиологии и патогенезе заболевания.
Семейный характер заболевания. Если врач при обследовании больного получает сведения о сходных случаях заболевания в семье, это прямо указывает на их возможную наследственную этиологию. В то же время заболевание только у одного члена родословной не исключает наследственного характера нарушения, поскольку оно может быть результатом новой доминантной мутации у одного из родителей или гетерозиготности обоих родителей по рецессивной болезни (сегрегация мутантного фенотипа).
Хроническое прогредиентное рецидивирующее течение - отличительный признак многих наследственных заболеваний. Болезни, начинающиеся в любом возрасте, имеют хроническое течение с прогредиентной клинической картиной.
Приведем несколько примеров. Хроническая пневмония с бронхоэктазами формируется у детей с легочной формой муковисцидоза. Длительные расстройства пищеварения возникают при целиакии (глютеновой энтеропатии), кишечной форме муковисцидоза и дисахаридазной недостаточности. Хронический процесс при наследственных болезнях развивается в результате постоянного действия мутантного гена. Хронизация, прогредиентность и рецидивирующее течение одного и того же заболевания по-разному выражены у различных больных, что объясняется взаимодействием генов между собой и с факторами внешней среды (генотип каждого человека индивидуален). К генетическим причинам относят особенности мутантных аллелей, а к средовым - активацию микробного фактора, нарушение питания и дополнительные повреждающие воздействия (охлаждение, инфекционные заболевания, стрессы).
Специфические симптомы наследственных болезней. Строго говоря, сугубо специфических симптомов наследственных болезней, вероятно, нет. Тем не менее редко регистрируемые симптомы, а особенно их сочетания, дают основание заподозрить наследственную этиологию заболевания. Например, вывих или подвывих хрусталика глаза характерен для синдрома Марфана, Вейля-Марчезани и гомоцистинурии. Голубые склеры обнаруживают при несовершенном остеогенезе и некоторых других болезнях соединительной ткани.
При алкаптонурии моча на пеленках темнеет. От больных с фенилкетонурией исходит мышиный запах. При кровоточивости можно предполагать болезнь фон Виллебранда или гемофилию. Грубые черты лица имеют больные с мукополисахаридозами. Астеническое телосложение с деформированной грудной клеткой характерно для синдрома Марфана. Непропорциональные конечности и туловище, низкий рост и своеобразный лицевой череп свидетельствуют об ахондроплазии. Право- и левосторонняя асимметрия размеров лица и конечностей позволяет предполагать наследственную гемигипертрофию.
Множественные патологические изменения органов и систем. Первичное вовлечение в патологический процесс множества органов или даже систем позволяет думать о наследственной этиологии заболевания. Большинство мутантных генов, вызывающих наследственные болезни, оказывают плейотропный эффект, в результате чего в процесс вовлекаются многие органы (подробнее о плейотропном действии гена - см. гл. 18).
Врожденный характер заболевания. И нормальные, и патологические аллели включаются в работу в разные периоды онтогенеза - от эмбрионального до старческого. Как подчеркивалось выше, врожденность патологических признаков не всегда свидетельствует о наследственной этиологии заболевания. Тем не менее более 25% всех моногенных наследственных болезней и почти все хромосомные нарушения начинают формироваться внутриутробно. Если ребенок рождается с комплексом патологических признаков, болезнь считают врожденной. Примеры врожденных наследственных болезней - хромосомные синдромы, ахондроплазия, Х-сцепленная гидроцефалия, аутосомно-рецессивная микроцефалия и др. Среди врожденных, но ненаследственных болезней выделяют краснушный, талидомидный, сифилитический, алкогольный, гидантоиновый и некоторые другие синдромы, этиологию которых устанавливают при целенаправленном сборе анамнеза, относящегося к первым неделям беременности. Врожденными нередко бывают наследственные болезни обмена веществ. Показания для биохимической и молекулярно-генетической диагностики болезней у младенцев: рвота, отказ от приема пищи, судороги, гипервентиляция, летаргия, кома, желтуха, гипертермия и измененный тонус мышц.
Резистентность к наиболее распространенным методам лечения. Одна из особенностей наследственных болезней - неэффективность лечения, хотя и не абсолютная. Это вполне понятно, потому что «исправить» первичные звенья, даже если известен первичный продукт мутантного гена, удается далеко не всегда (мукополисахаридозы, миодистрофия Дюшенна, нейрофиброматоз). Естественно, что толерантность к лечению свойственна не всем болезням. Если расшифрованы ключевые звенья патогенеза, то возможна разработка успешных методов лечения. Некоторые заболевания из группы устойчивых к терапии переходят в группу поддающихся лечению (гепатолентикулярная дегенерация, целиакия, муковисцидоз).
Клинико-генеалогический метод
Генеалогией в широком смысле слова называют родословную. Генеалогический метод - метод родословных, т.е. прослеживание болезни (признака) в семье или в роду с указанием типа родственных связей между членами родословной. В медицинской генетике этот метод называют клинико-генеалогическим.
Генеалогический метод - один из наиболее универсальных в медицинской генетике. Его широко применяют в целях медико-генетического консультирования для установления наследственного характера признака, определения типа наследования и пенетрантности гена, анализа сцепления генов и картирования хромосом, изучения интенсивности мутационного процесса и расшифровки механизмов взаимодействия генов.
Эмпирические наблюдения родословных с наследованием патологических признаков известны давно. Например, в Талмуде отражено понимание сцепленного с хромосомой Х наследования гемофилии. В середине XVIII в. П. Мопертюи описал наследование доминантного признака - полидактилии - и правильно проанализировал его расщепление в потомстве. В начале XIX в. Дж. Адамс на основе эмпирического анализа родословных описал доминантный и рецессивный типы наследования. Несколько врачей подробно разобрались в наследовании гемофилии и цветовой слепоты. Эти и некоторые другие попытки анализа родословных можно рассматривать как предпосылки формирования генеалогического метода, которое закончилось в начале XX в., вскоре после рождения генетики как науки. С этого времени метод широко используют в генетике человека и медицинской генетике. Его дальнейшее усовершенствование шло по линии составления родо словных, и особенно разработки методов статистического анализа данных. Метод находил все более широкое применение в клинической генетике и генетике человека (изучение мутационного процесса, сцепления генов и др.).
Суть генеалогического метода сводится к прослеживанию признака или болезни среди близких и дальних прямых и непрямых родственников. Технически он складывается из двух этапов: составления родословной и генеалогического анализа.
Составление родословной. Сбор сведений о семье начинают с консультирующегося или пробанда. Консультирующимся называют человека, обратившегося к врачу. Это не обязательно больной. Пробанд - больной или носитель изучаемого признака. Во многих случаях консультирующийся и пробанд - один и тот же человек. Детей одной родительской пары называют сибсами (братья и сестры). Название «сибс» происходит от английской аббревиатуры SIBS: SIsters-BrotherS. Ядерной семьей в узком смысле называют родительскую пару и их детей. Обычно родословную собирают по одному или нескольким признакам, обращая особое внимание на родственные связи больных с определенным заболеванием, по поводу которого семья обратилась за медико-генетической консультацией.
В зависимости от цели исследования родословная может быть полной или ограниченной. Желательно стремиться к наиболее полному составлению родословных по восходящему, нисходящему и боковым направлениям. Эта задача не так легка, как может показаться на первый взгляд. Чем больше поколений вовлечено в родословную, тем она обширнее.
Составление родословной сопровождают краткой записью о каждом ее члене с точной характеристикой его родства с пробандом (легенда родословной). В дальнейшем для наглядности или при публикации родословную изображают графически. Для этого обычно используют стандартные символы.
При применении генеалогического метода в родословной важно отмечать обследованных на присутствие признака (лучше использовать сведения о заболевании из объективного источника, например из истории болезни) и необследованных, сведения о которых почерпнуты из ответов пробанда, родственников или из анкет. Грубая ошибка - искусственное укорочение звеньев родословной вследствие трудностей обследования родственников II и III степени родства, особенно если не указано, у кого из членов родословной действительно не было родственников, а у кого сведения не собраны.
Получить сведения о родственниках непросто. Во-первых, не все пациенты знают об их болезнях. Во-вторых, они нередко скрывают семейные случаи из-за ложного стыда или, наоборот, «открывают» их у родственников супруга, стараясь свалить на них вину за болезнь ребенка. Для получения семейных сведений можно применять анкетирование. При правильном составлении перечня вопросов и доступности формулировок для понимания членами семьи, не имеющими медицинского образования, анкетирование дает достаточно полную информацию. Очень важно при необходимости провести личный осмотр и дополнительное обследование родственников больного.
Одна из распространенных ошибок применения генеалогического метода - ограничение анализа только опросом родственников (или о родственниках). Даже подробного опроса, как правило, недостаточно. Некоторые члены родословной часто нуждаются в полном клиническом, параклиническом или лабораторногенетическом (цитогенетическом, биохимическом и др.) обследовании, что требует дополнительных расходов. План такого обследования необходимо тщательно рассмотреть с генетической точки зрения в соответствии с принципом «меньше нельзя, а больше не нужно».
Помощь клинико-генеалогического метода в диагностике наследственных нарушений очевидна. Так, если в родословной обнаружено наследование болезни, соответствующее определенному типу (доминантному, рецессивному или Х-сцеплен ному), это позволяет укрепиться в предположении о ее наследственной этиологии, а в некоторых случаях помогает и в диагностике.
Генеалогический анализ. Первая задача при анализе родословной - установление наследственного характера признака. Если в родословной один и тот же признак (болезнь) встречается несколько раз, можно думать о его наследственной этиологии, но прежде всего необходимо исключить возможность фенокопии. Например, если патогенный фактор действовал на женщину во время всех беременностей, то могут родиться несколько детей с врожденными пороками. Другой пример: одни и те же профессиональные вредности или внешние факторы могут вызывать сходные заболевания у членов одной семьи.
Если действие внешних факторов исключено (для разных поколений оно исключается с большей вероятностью), говорят о наследственном характере болезни. С помощью генеалогического метода было открыто большинство наследственных заболеваний.
Вторая задача - установление типа наследования при обнаружении наследственного характера признака (болезни). Подробнее об установлении типа наследования по родословной сказано в гл. 5.
Проблемы диагностики в клинической генетике
Точная идентификация наследственного заболевания (диагностика) затруднена прежде всего в результате отсутствия при большинстве наследственных заболеваний (за редким исключением) патогномоничных признаков. Диагностика наследственных болезней также осложняется их редкостью, что затрудняет приобретение собственного опыта в этой работе.
Чаще всего один и тот же патологический признак обнаруживают при нескольких и даже многих формах наследственных болезней. Основные сложности в диагностике врожденных и наследственных нарушений связаны с их генетической гетерогенностью, под которой понимают сходство клинических признаков, обусловленное мутациями различных генов (например, высокий рост при болезни Марфана, гомоцистинурии, синдроме Клайнфелтера и других марфаноподобных синдромах). Диагностика наследственного заболевания также осложнена фенотипическим (клиническим) полиморфизмом нарушений, когда при одной и той же унаследованной генной мутации могут развиться как ее ярко выраженные, так и стертые или даже различные клинические формы (патологические фенотипы). Так, муковисцидоз может манифестировать у новорожденных как мекониальный илеус, хронический воспалительный процесс в легких у детей более старшего возраста или синдром нарушенного кишечного всасывания. Другой пример: миопатии Дюшенна и Беккера - генетически единая форма, клинически относящаяся к разным болезням.
Осложняет диагностику существование фенокопий наследственных болезней - приобретенных заболеваний с идентичными симптомами. Эту так называемую имитацию менделизма можно продемонстрировать на примере сходства синдрома Вильямса (аортальный стеноз, «лицо эльфа», задержка умственного и физического развития) с краснушной эмбриопатией. Фетальный алкогольный синдром может возникать у нескольких сибсов в семье, имитируя наследственную микроцефалию с лицевыми дизостозами.
На самом деле реальные, четко выраженные фенокопии регистрируют нечасто.
Трудности диагностики наследственных заболеваний также связаны с существованием некоторых генетических явлений, оказывающих существенное влияние на формирование клинического фенотипа, таких как мозаицизм, экспансия аллелей, однородительское наследование (дисомия и изодисомия) и геномный импринтинг.
Проблемы дифференциальной диагностики в клинической генетике
При дифференциальной диагностике наследственного заболевания на основании клинических признаков, вероятно, можно использовать обобщающий подход, близкий к алгоритмической процедуре. Он представлен рядом ступеней.
Составляют перечень клинических признаков у конкретного больного.
На основе описанных признаков составляют перечень возможных сходных заболеваний.
Составляют описание признаков для каждого из заболеваний.
Сравнивают клинические признаки у конкретного больного и симптомы заболевания А. Если окажется, что существенных общих признаков мало, то у пациента не болезнь А. Переходят к следующему пункту. Если существенные признаки общие, то у больного заболевание А. Алгоритм остановлен.
Сравнивают признаки заболевания у пациента с признаками болезни Б. Если окажется, что существенных общих признаков мало, то у него не болезнь Б. Переходят к следующему пункту. Если существенные признаки общие, то у пациента болезнь Б. Алгоритм остановлен.
Продолжают до тех пор, пока не будет установлена тождественность признаков болезни у пациента с одним из дифференцируемых заболеваний.
Назвать такой способ дифференциальной диагностики алгоритмом нельзя, так как он, являясь массовым, не характеризуется определенностью. Действительно, как составить по этому перечню признаков весь спектр сходных заболеваний? Как разделить существенные и несущественные признаки и откуда их взять? Как определить, что симптомы носят общий существенный характер? Если бы удалось вполне определенно ответить на поставленные вопросы, то описанный подход, вероятно, можно было бы назвать алгоритмом. Алгоритмы в дифференциальной диагностике возникают тогда, когда составлена точная опись признаков болезней и количественно оценена вероятная доля каждого из них, т.е. когда на основе статистического анализа будут установлены количественные критерии для симптомов сходных заболеваний.
Диагностика, в том числе и дифференциальная, может быть успешной, если она основана на достоверных и объективных данных. Для создания диагностических программ необходимы, с одной стороны, детальная обработка медицинских данных и их представление в формализованном виде, а с другой - сведения о частоте и удельном весе каждого из признаков заболевания. Именно поэтому большое значение приобретает соблюдение принципа группировки информации о пациенте и его заболевании. Всю информацию, получаемую врачом или исследователем о больном, можно классифицировать по нескольким принципам.
Информация по достоверности:
Информация по диагностическому значению:
Необходимой и патогномоничной информацией считают, например, ЭКГприз нак - удлинение интервала Q-T при синдроме удлиненного интервала Q-T. Специфическая патогномоничная информация служит предпосылкой достоверного диагноза с использованием условно категорического или простого категорического силлогизма.
Условно специфическая информация (желтуха, рвота, отеки, эритроцитурия и др.), а также неполные специфические синдромы служат предпосылками для дифференциальной диагностики и установления вероятного диагноза.
Неспецифическая информация не имеет большого диагностического значения. Она служит следствием патологического процесса, лежащего в основе болезни, но отражает общие, неспецифические реакции организма. К ним относят слабость, быструю утомляемость, снижение аппетита, похудание, головную боль, плохой сон и др.
Случайная информация - признаки, не имеющие отношения ни к основному, ни к сопутствующим заболеваниям: рвота при введении лекарственного средства, его непереносимость, тахикардия при волнении, обморок при виде крови и др.
Деление это условно, так как один и тот же симптом или лабораторный показатель при одной болезни может быть условно специфическим, при другой - неспецифическим, при третьей - второстепенным или случайным. Он может входить в состав специфического наследственного синдрома. Именно поэтому оценку диагностического значения информации (симптомов, синдромов, показателей) следует проводить применительно к каждой болезни и даже к ее отдельным фазам.
Компьютерные программы диагностики наследственных заболеваний. Огромный спектр наследственных болезней и практическая потребность в диагностике их многочисленных форм и врожденных пороков развития невозможны без сравнительного анализа данных литературы. В связи с этим на протяжении последних 20 лет стали создавать компьютерные информационные базы данных и диагностические программы. Их назначение - ускорение и объективизация выбора диагноза из множества генетически разнородных, но клинически сходных синдромов и болезней.
Медико-генетическая консультация и необходимость оказания высокоэффективной медико-генетической помощи при наследственных болезнях требуют точного диагноза. В связи с тем что генетические нарушения редки, большинство врачей и даже медицинских генетиков имеют собственный опыт только по нескольким случаям конкретной болезни. Это диктует необходимость постоянного знакомства врачей с описанными в литературе случаями генетических дефектов, что позволяет установить точный диагноз, а также своевременно дать полноценный медикогенетический прогноз для родителей и семьи пробанда.
Каталоги необходимы для практической работы клинических генетиков. Они обеспечивают врача информацией или служат гидами к данным по диагнозу, лечению или генетическому консультированию. Поиски по отдельным словам или их комбинациям могут генерировать перечень форм, в которых эти слова или комбинации можно обнаружить. Это хорошее начало для поиска диагноза заболевания, позволяющее назначить правильное лечение и провести консультирование конкретной семьи.
Общий принцип диагностики с использованием компьютерных программ заключается в том, что симптомы, обнаруженные врачом, вводят в компьютер и на их основе осуществляют компьютерный поиск наиболее вероятных диагнозов. После этого врач может обратиться за справкой в базу данных по выбранным диагнозам и получить описание синдрома (болезни) и даже фотографии больных. Таким образом, врач принимает решение о диагнозе и выбирает способ его верификации, если в этом есть необходимость.
В зарубежной литературе есть несколько каталогов и баз данных, содержащих подробную информацию о наследственных болезнях, генах и хромосомах человека. Авторы этих каталогов - ведущие медицинские генетики V.A. McKusick (США), M. Baraitser, R. Winter (Великобритания), D. Borgaonkar (США), J. Fresal (Франция), A. Schinzel (Швейцария) и T.B. Pitt (Австралия).
Компьютерная диагностика позволяет использовать накопленный опыт и научные знания о манифестации патологического процесса на разных этапах его развития. В компьютерной памяти могут храниться сведения о клинических признаках и результатах обследования при самых редких нарушениях. В настоящее время клиницисты-генетики не должны работать без справочно-диагностических систем.
Приводим наиболее известные компьютерные информационные базы данных и диагностические программы.
Менделирующая наследственность человека (Mendelian Inheritance in Man. V.A. McKusick, MIM). Интернет-версия «On Line Mendelian Inheritance in Man - OMIM (адрес в Интернете - www.ncbi.nlm.nih.gov) - информационнопоисковая мультимедийная система. Каталог содержит более 13 500 статей, и базу его данных ежедневно пополняют. В каталоге представлены данные о молекулярных дефектах при менделирующих заболеваниях (перечень нарушений и продуктов патологических генов), генетические карты человека (аутосомные, Х- и Y-хромосомные, митохондриальные), описание каталогов аутосомнодоминантных, аутосомно-рецессивных, Х- и Y-хромосомных генов, каталог митохондриальных генов, список литературы и предметный указатель. Каждый менделирующий признак имеет шестизначный номер (например, фенилкетонурия - MIM 261600). За названием признака и синонимами следует лаконичное описание болезни и ее генетика. Статья заканчивается списком наиболее важных источников литературы по теме. Каталог снабжен полным предметным указателем.
Оксфордская медицинская база данных (Oxford Medical Database - OMD) имеет два подраздела - лондонскую базу данных (London Dysmorphology Database - LDDB), включающую сведения более чем о 2300 нехромосомных синдромах, и лондонскую нейрогенетическую базу данных (London Neurogenetics Database - LNDB), содержащую сведения более чем о 2200 наследственных заболеваниях центральной и периферической нервной системы. Обе базы были созданы M. Baraitser и R. Winter в качестве инструментов для клинической диагностики врожденных аномалий и нейрогенетических синдромов соответственно. Адрес OMD в Интернете: www.dhmhd.mdx.ac.uk. Базы данных основаны на сведениях более чем из 1000 научных журналов и постоянно пополняются. Их создание было обусловлено необходимостью диагностики и обеспечения генетического консультирования при сотнях редких синдромов. Оба автора - опытные клинические генетики, и система во многом основана на их клинической практике и опыте. Работа с базой данных требует практических навыков, потому что она в большей степени - система для экспертов, чем экспертная система. Она очерчивает границы для опытного клинициста, способного сопоставить важные клинические признаки и оценить сходство с таковыми, описанными в литературе. Фотобиблиотека введена в 1993 г.; версию для Windows открыли в 1996 г.
В результате четкого описания симптомов, обнаруживаемых у конкретного больного, указанные диагностические системы и базы данных позволяют из всего многообразия заболеваний выделить наиболее сходные по фенотипическим признакам синдромы. Врач получает возможность провести дифференциальную диагностику среди ограниченного перечня нозологических форм, выбрать оптимальный план дальнейшего обследования и лечения больного или использовать информацию для оценки генетического риска (прогноза заболевания). Возможности диагностики наследственных заболеваний увеличиваются при обеспечении обмена информацией через электронные средства связи между всеми пользователями этих систем.
Международное признание получила австралийская компьютерная система идентификациии наследственнных синдромов - POSSUM (Pictures of Standrad Syndromes and Undiagnozed Malformations). Каждый из идентифицированных синдромов сопровождается рядом стандартных фотографий и описанием симптомов. Окончательный диагноз основан на сравнительной оценке изображения больного и фотографии.
Существует Марсельская система диагностики наследственных болезней GENDIAG (Франция), основанная на базе данных по симптомам и результатам функциональных проб. Принцип ее работы базируется на предположении о том, что отклонения в развитии организма или органов можно рассматривать как список симптомов, с каждым из которых связан индикатор частоты манифестации конкретного признака. Идентификацию в системе осуществляют при обращении к базе данных, включающей сведения о 3000 признаках и результатах функциональных проб, организованных в иерархическую последовательность. Диагноз отвергают, если у пациента обнаруживают признаки, не перечисленные в списке симптомов рассматриваемой нозологической формы.
Атласы по врожденной и наследственной патологии. Большую помощь в диагностике наследственных болезней можно получить из атласов, в которых приведены фотографии больных (с акцентом на основные симптомы болезни), описание диагностических признаков, а также предметный указатель по симптомам и болезням. Ниже приведен список наиболее удачных атласов.
Аномалии развития (иллюстрированное пособие для врачей) / Под общ. ред. В.В. Кра силь никова. - СПб.: ФОЛИАНТ, 2007. - 336 с.
Беляков Ю.А. Наследственные болезни и синдромы в стоматологической практике. - М.: Медицина, 2008. - 238 с.
Джонс Л. Наследственные синдромы по Дэвиду Смиту. Атлас-справочник. - М.: Практика, 2011. - 1022 с.
Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. - М.: Товарищество научных изданий КМК, 2007. - 448 с.
Мордовцев В.Н., Мордовцева В.В., Мордовцева В.В. Наследственные болезни и пороки развития кожи. Клиника. Морфология. Лечение. Атлас. - М.: Наука, 2004. - 174 с.
Hennekam C.M., Krantz D., Allanson Gorlin J.E. Syndromes of the Head and Neck. - Oxford: Oxford Press, 2010. - 1452 p.
Синдромологический подход к диагностике
Точная и ранняя диагностика наследственных болезней имеет важнейшее значение как для своевременного назначения полноценного лечения, так и для эффективного медико-генетического прогноза и предупреждения повторных случаев заболевания в семье. Поиски диагностических критериев наследственных нарушений были направлены на определение патогномоничных клинических или лабораторных признаков заболеваний. Несмотря на многолетние и многочисленные исследования, такие признаки были найдены лишь для единичных заболеваний (например, удлиненный интервал Q-T при синдроме удлиненного интервала Q-T, симптом кольчатых волос при синдроме с аналогичным названием, узелки Лиша при нейрофиброматозе 1-го типа, кольцо Кайзера-Флейшера при болезни Вильсона-Коновалова).
Таким образом, ориентация только на специфические признаки остается нереальной задачей и не решает проблему диагностики наследственных заболеваний. Клинические признаки подобных нарушений не отличаются специфичностью, и лишь их определенные сочетания служат опорой для дифференциальной диагностики как в группе сходных наследственных болезней, так и среди заболеваний ненаследственной этиологии. Многие наследственные нарушения носят характер синдромов. Частоту синдромной манифестации можно объяснить развитием заболеваний, исходящих из определенных зародышевых листков. Так, частое сочетание поражения нервной системы и кожи может быть связано с развитием обеих систем из эктодермы.
В связи с этим большое значение приобретает проблема выделения минимального диагностического симптомокомплекса обязательных признаков, а также необходимость учета отрицательных симптомов, исключающих диагноз. При его установлении предполагают соответствие типичной нозологической форме. Известно, что на этом этапе диагностики редких форм наследственных болезней существуют объективные сложности, не зависящие от уровня знаний и умений практического врача. Для этого необходимо выделить комплекс обязательных признаков или ядро синдрома, отделить его от менее распространенных дополнительных признаков, а также от осложнений и случайных сочетаний.
Возможности нозологической дифференциации на основе клинико-лаборатор ного анализа без патогномоничных лабораторных признаков можно представить на примере одной из форм органической ацидурии - метилмалоновой ацидемии.
На первом этапе изучения заболевания проводили анализ признаков по опубликованным описаниям больных. Из литературы отбирали случаи по присутствию триады симптомов: рвота, летаргия, токсикоз. Эти признаки отмечены у больных в первых сообщениях, датированных ХХ в., хотя описания публиковали под другими названиями болезней. Необходимо было проанализировать характер и детали этих признаков, частоту их обнаружения у больных в последующих сообщениях, а также выяснить возможное существование других постоянных признаков этого заболевания. По указанной триаде были отобраны случаи для фенотипического анализа. Затем в этой группе с учетом типа наследования заболевания проводили дополнительный комплекс исследований. Была установлена 100% частота обнаружения трех признаков. Кроме того, отмечена высокая распространенность поражения слизистых оболочек, кожи и др.
На втором этапе была проанализирована клиническая картина у больных в собственных исследованиях и наблюдениях. У них обнаруживали широкий спектр признаков болезни. Среди них отмечали ряд постнатальных отклонений, а также патологические изменения внутренних органов - почек, кожи и нервной системы. У больных регистрировали нарушения нервной системы: задержку речевого, психомоторного развития, судороги, астеноневротический синдром и изменения на электроэнцефалограмме. При многопрофильном обследовании больных, направленных в консультативные учреждения с диагнозом «перинатальная энцефалопатия», вышеупомянутые симптомы рассматривали как симптомы или осложнения энцефалопатии. К последним относили не только обычные симптомы, сопутствующие заболеванию, но и более редкие изменения - помутнение роговицы и олигофрению.
Эти вторичные изменения, возникающие, вероятно, вследствие неполноценного функционирования митохондрий, не считают специфическими и до установления диагноза не рассматривают в качестве ее симптомов. Они по-разному выражены у больных и имеют возрастную динамику. Что касается отдельных симптомов, то они не имеют специфического характера, подвержены возрастной динамике, но в комплексе и на фоне других дефектов образуют характерный для метилмалоновой ацидемии признак.
Таким образом, выше описан процесс формирования синдромологического подхода к диагностике наследственного заболевания на основе глубокого анализа всех симптомов, обнаруживаемых у больных, и сопоставления их диагностической значимости.
Поскольку до сих пор практически не обнаружены патогномоничные признаки наследственных заболеваний, врачу чаще приходится сталкиваться с ситуацией, когда один и тот же симптом обнаруживают при нескольких и даже многих формах нарушений. Например, более 150 форм наследственных заболеваний характеризуются симптомокомплексом нарушения слуха; более чем при 30 наследственных болезнях обнаруживают деформации грудной клетки в виде воронки или киля. Искривление позвоночника регистрируют более чем при 50 наследственных синдромах. Аномалии почек известны при 30 синдромах.
Чрезвычайно редко наследственные болезни манифестируют только моносимптомом. Именно поэтому задача врача состоит в том, чтобы обнаруживать при клиническом осмотре дополнительные признаки, существенно облегчающие дифференциальную диагностику. Например, при существовании у больного врожденного порока сердца необходим тщательный осмотр кистей верхних конечностей, так как обнаружение укорочения I пальца кисти или трех его фаланг вместо двух наводит на предположение о доминантно наследуемом синдроме Холта-Орама (синдром «рука-сердце»). Больные с этим синдромом часто бывают пациентами кардиологических отделений и клиник сердечно-сосудистой хирургии.
Резко выступающие надбровые дуги могут быть признаком синдрома фронтометафизарной дисплазии (Х-сцепленная форма остеодисплазии Мелника-Нидлса), а запавшая переносица - мукополисахаридозов или ахондроплазии.
Искривление нижних конечностей рахитического типа - результат не только классического рахита, как полагали ранее, но может быть следствием нарушения обмена в костях или процессов роста и развития хряща, отмечаемых при более чем 40 различных наследственных болезнях.
Огромное число наследственных заболеваний (более 300) сопровождается поражением кожи и ногтей (гипоплазию или дисплазию регистрируют более чем при 20 генетически обусловленных болезнях). Синдромологический подход к диагностике наследственных форм патологических изменений в дерматологии особенно значим, как ни в какой другой области медицины.
Наследственные синдромы с вовлечением в процесс зубов постоянно встречаются в практике стоматологов. Не менее 20 наследственных синдромов сопровождаются изменением зубов (дистрофия эмали, неправильная форма зубов, сверхкомплектность, раннее выпадение, множественный кариес и др.). Дефекты зубочелюстной системы нередко обнаруживают при мукополисахаридозах, синдроме Элерса-Данло, пахионихии, фосфат-диабете, синдроме де Тони-Дебре-Фанкони и многих других.
Для педиатрической и медико-генетической практики осмотр детей, страдающих задержкой нервно-психического развития, - повседневная практика. Этот синдром обнаруживают при многих наследственных болезнях обмена веществ, хромосомных аномалиях, болезнях накопления, митохондриальных заболеваниях и более чем при 150 наследственных синдромах. Задержка психоречевого и моторного развития - прямое показание для обязательного исключения наследственной патологии у пациента.
Следует подчеркнуть, что для диагностики наследственных и врожденных заболеваний можно клинически обнаружить более двухсот внешних симптомов или отдельных признаков без привлечения дополнительных методов исследования. Нужно только стремиться к их поиску.
Естественно, что, в зависимости от профиля клиники, отдельные симптомы будут регистрировать чаще. В офтальмологическом отделении чаще будут обнаруживать симптомы поражения органа зрения, в ЛОР-клинике - нарушения слуха, речи и др. Именно поэтому чем менее распространено заболевание, сопровождающееся поражением какого-либо органа или системы, тем более вероятно, что его будут обнаруживать в специализированном учреждении.
Так, больных с синдромом Марфана (заболеваемость - 1:15 000) и гомоцистинурией (частота - 1:50 000) можно обнаружить в глазных (вследствие высокой миопии и подвывиха хрусталика) и хирургических (по поводу деформации грудной клетки) клиниках. Больные низкого роста чаще будут попадать в поле зрения эндокринолога, с поражением суставов - на прием ревматолога или артролога и др.
Таким образом, чем большими знаниями в области наследственных нарушений и синдромологии обладает узкий врач-специалист, тем быстрее будет установлен диагноз или больной направлен к врачу-генетику. Поскольку многие наследственные заболевания и синдромы регистрируют редко, знание узкими специалистами этого небольшого числа нозологических форм крайне необходимо. Диагностику синдромных нарушений могут облегчить разрабатываемые генетические регистры и компьютерные диагностические системы, специализированные по узкому профилю.
ДИАГНОСТИЧЕСКОЕ ЗНАЧЕНИЕ СПЕЦИФИЧЕСКИХ СИМПТОМОКОМПЛЕКСОВ В ПРОЦЕССЕ ВЕРИФИКАЦИИ НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ
В развитии наследственных признаков или болезней принимает участие как наследственная конституция (генотип), так и внешняя среда. Действие любых генов осуществляется в тесном взаимодействии с факторами среды. Хотя для развития одних признаков или болезней определяющую роль играет наследственность (генотип), а для других существенное значение имеет внешняя среда, нет таких признаков, которые зависели бы только от наследственности или только от среды. При различных условиях среды может быть разная степень экспрессии гена и, следовательно, различная выраженность фенотипа.
Симптомы врожденных и наследственных болезней очень разнообразны. В этом разделе представлена лишь часть наиболее значимых симптомов, которые регистрируются в медико-генетической практике и нередко вызывают диагностические трудности.
МакроцефалияМакроцефалия (макро- + греч. kephalē - голова; син. макрокефалия, мегалокефалия, мегацефалия) - аномалия развития, характеризующаяся чрезмерно большой головой. О макроцефалии следует говорить при увеличении ее размеров более двух стандартных отклонений для соответствующего возраста (примерно 5 см и более). Этот симптом обнаруживают при многих наследственных и ненаследственных заболеваниях.
При оценке размеров головы необходимо проводить сравнение ее окружности у конкретного больного с возрастными нормативами, желательно определенными для данного региона и этнического состава популяции. К сожалению, подобные нормативы мало разработаны или разработаны для отдельных регионов, и в практической работе в большинстве случаев используют антропометрические среднестатистические показатели, утвержденные органами здравоохранения.
Для правильной интерпретации данных окружности головы и объема черепа необходимо использовать центильные таблицы соответствующих параметров в зависимости от возраста детей. Сравнение индивидуальных показателей с соответствующими центильными шкалами позволяет определить степень отклонений параметров от возрастной нормы. Количественная оценка позволяет не только сравнить их, но и получить объективную характеристику размеров головы обследуемого ребенка.
В оценке макроцефалии как симптома необходимо учитывать возможность физиологического увеличения черепа, когда лицевой и мозговой череп находятся в соответствующей пропорции и форма черепа не изменена, а он лишь увеличен в размере. У недоношенных детей макроцефалия связана с относительной незрелостью и диспропорцией между головой и туловищем. Восстановление пропорций происходит к 12-15-му месяцу жизни, при этом какие-либо неврологические или рентгенологические патологические изменения отсутствуют.
Существуют семейные формы (доминантное или рецессивное наследование) увеличения размеров головы, но, даже исключая их, около 7-10% детей имеют размеры, отличающиеся от нормы более чем на два стандартных отклонения. Именно поэтому при нормальных показателях неврологического, психического и интеллектуального развития, а также рентгенологического исследования при гармоничном увеличении размеров черепа проводить более углубленное обследование пациента нецелесообразно. Важнейший критерий патологического состояния - динамическое наблюдение за ребенком. Родители должны получить рекомендации приходить на контрольные осмотры 1 раз в полгода. Вместе с тем существует множество патологических состояний (в том числе наследственных и врожденных), при которых отмечают увеличение окружности головы (макро- и мегалоцефалию).
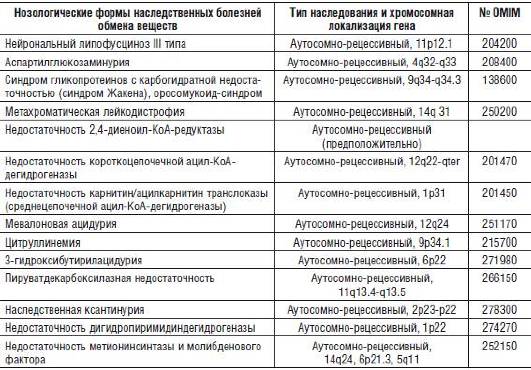
Макроцефалию как симптом патологических изменений можно обнаружить при врожденных пороках развития костей черепа, а также при ряде наследственных нарушений обмена веществ (табл. 15-1).
Таблица 15-1. Основные формы наследственных болезней обмена веществ, сопровождающихся макроцефалией
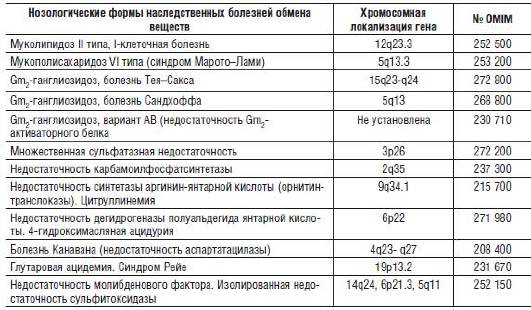
Большую группу заболеваний составляют наследственные синдромы, сопровождающиеся макроцефалией (табл. 15-2).
Таблица 15-2. Некоторые наследственные синдромы, сопровождающиеся макроцефалией
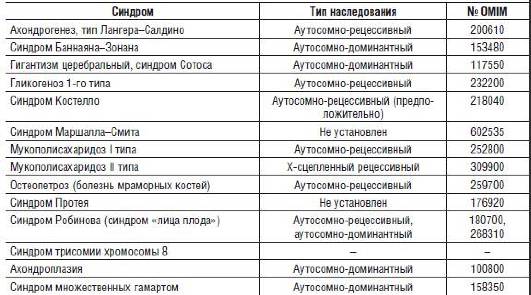
Микроцефалия Микроцефалия (микро- + греч. kephalē - голова) - аномалия развития, характеризующаяся малыми размерами головного мозга и мозгового черепа; уменьшение размеров черепа (головы) ниже возрастной нормы.
О микроцефалии говорят, если размеры окружности головы уменьшены на 10% и более по сравнению с возрастной нормой.
В психоневрологии под этим термином часто понимают тяжелые дегенеративные заболевания ЦНС, сопровождающиеся уменьшением окружности головы и объема головного мозга при отсутствии внутричерепной гипертензии. При подобных заболеваниях часто одновременно отмечают раннее закрытие родничков и формирование небольших деформаций черепа, имеющих сходство с формой черепа при краниостенозах. Напротив, ряд последних сопровождается уменьшением размеров головы (особенно при раннем закрытии нескольких швов). В такой ситуации важно понимать этиологию уменьшения размеров головы. При микроцефалии рост костей снижен, но он не остановлен, и поэтому синостоз костей черепа происходит медленно, в то время как при краниостенозе закрытие швов наблюдают уже при рождении. Особенно отчетливо этот признак заметен на рентгенограммах костей черепа. При микроцефалии часто существует задержка психомоторного развития, связанная с повреждением мозга и диагностируемая на ранних этапах жизни ребенка. Признаки повреждения мозга и отсутствие внутричерепной гипертензии обнаруживают при компьютерной или магнитнорезонансной томографии головного мозга.
Анализ, казалось бы, такого простого симптома, как микроцефалия, показывает, что ошибочная трактовка этиологии этого признака может серьезно повредить больному. При развитии раннего синостоза черепных швов и уменьшении прироста окружности головы с некоторой задержкой психомоторного развития ребенка причину отставания умственного развития можно устранить путем нейрохирургической коррекции краниостеноза. При микроцефалии хирургическое вмешательство не показано.
Выделяют истинную микроцефалию (синдром Джакомини) и микроцефалию, наследуемую по аутосомно-рецессивному или рецессивному, сцепленному с Х-хромосомой типу.
Кроме того, микроцефалия возникает при многих наследственных синдромах и наследственных заболеваниях обмена веществ. Иногда регистрируют семейную микрокранию (у нескольких членов семьи) или физиологическую микроцефалию. Эти состояния не сопровождаются неврологическими нарушениями.
Наиболее распространенные в медико-генетической практике наследственные синдромы, при которых обнаруживают микроцефалию, представлены в табл. 15-3.
Таблица 15-3. Некоторые наследственные синдромы, при которых обнаруживают микроцефалию
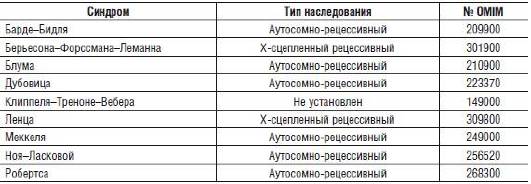
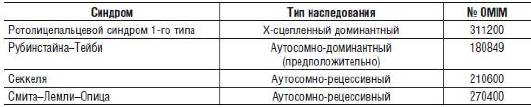
Микроцефалия может быть симптомом ряда наследственных болезней обмена веществ (табл. 15-4).
Таблица 15-4. Микроцефалия при ряде наследственных болезней обмена веществ
Многие хромосомные аномалии сопровождаются микроцефалией: синдром хромосомы 4р-; синдром хромосомы 9р+; синдром хромосомы 10q+; синдром Патау, синдром хромосомы 21q- и др.
Необходимо помнить, что к микроцефалии могут привести воздействия внешних средовых факторов: инфекционные заболевания (токсоплазмоз, краснуха, цитомегалия), прием алкоголя (фетальный алкогольный синдром), медикаментов (гидантоиновый синдром) и других соединений. Микроцефалия развивается при воздействии ионизирующего излучения в критические периоды внутриутробного развития (радиационный синдром, лучевая микроцефалия).
Нарушения нервно-психического развития Интенсивное развитие нервной системы происходит еще во время внутриутробной жизни, но ее анатомическое и функциональное совершенствование продолжается в постнатальном периоде.
Популяционная частота нарушений умственного развития у детей достигает 10%, но в большинстве случаев они представлены легкими и среднетяжелыми формами. У одного-двух из 10-15 умственно отсталых детей она достигает степени глубокой инвалидности. По обобщенным данным зарубежных авторов, распространенность нарушений умственного развития в детском возрасте в среднем составляет около 3% (Шапиро Б.К., 2004). Пик диагностики умственной отсталости приходится на возраст начала школьного обучения.
Обобщенные сведения о причинах нарушений нервно-психического развития детей в свете достижений современной генетики представлены в табл. 15-5.
Таблица 15-5. Структура этиологических факторов интеллектуальных нарушений у детей (обобщенные данные)
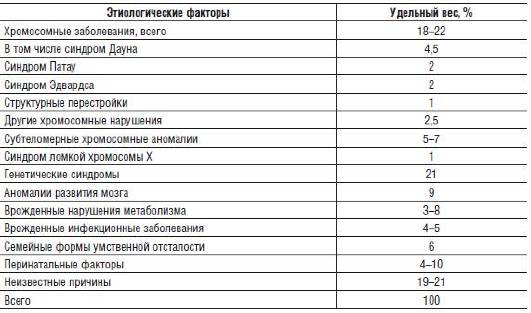
Как видно из табл. 15-5, несмотря на внедрение современных технологий диагностики нарушений развития плода, более чем в 20% случаев их этиология остается не идентифицированной, а хромосомные аномалии занимают ведущее место среди причин.
Таким образом, нарушения умственного развития в большинстве случаев могут быть обусловлены:
Олигофрения как психическое, и главным образом умственное, недоразвитие может быть связана с задержкой развития мозга в пренатальном периоде, нарушениями миграции нейронов, малыми аномалиями мозга, микроили макроцефалией, гидроцефалией и сложными (комбинированными) пороками развития.
Ультразвуковое сканирование плода дает возможность диагностировать пороки развития мозга, анэнцефалию, микро- и гидроцефалию, а также голопрозэнцефалию. Разрешающие способности метода не позволяют проводить диагностику на ранних этапах внутриутробного развития. В то же время техника УЗИ постоянно совершенствуется, внедряются различные технологии допплерометрии и магнитно-резонансной спектроскопии мозга, что расширит диагностические возможности уже в ближайшем будущем.
Тяжелые поражения мозга с олигофренией могут быть обусловлены резусконфликтом (гемолитическая болезнь новорожденных и билирубиновая энцефалопатия) и изоиммунизацией по тромбоцитарным антигенам. Это одна из причин внутричерепных кровоизлияний у новорожденных.
В ходе диагностического процесса важно обращать внимание на сочетания отклонений в умственном развитии с поражением других органов и систем. Наиболее значимые подобные ассоциации приведены в табл. 15-6-15-11.
Таблица 15-6. Сочетание задержки умственного развития с высоким ростом
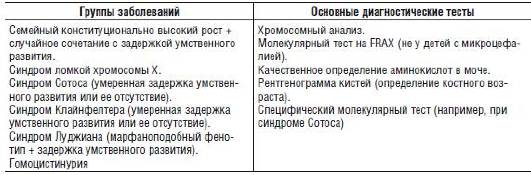
Таблица 15-7. Сочетание задержки умственного развития и макроцефалии
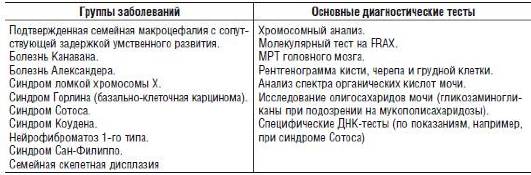
Таблица 15-8. Сочетание задержки умственного развития и микроцефалии
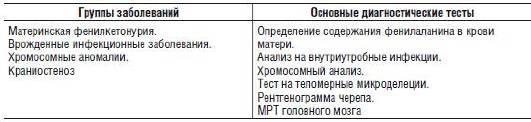
Таблица 15-9. Сочетание задержки умственного развития и ожирения
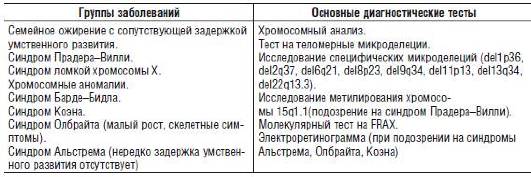
Таблица 15-10. Сочетание задержки умственного развития с гепатомегалией без значительной спленомегалии
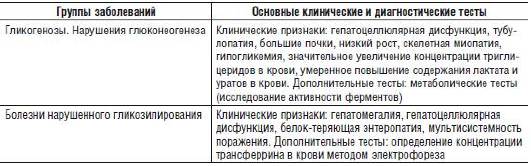
Таблица 15-11. Сочетание задержки умственного развития с гепатомегалией и спленомегалией
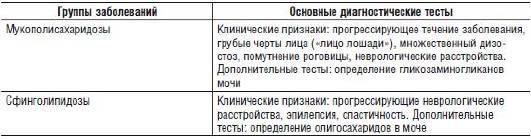
Нарушения зрения
Среди всех форм глазных заболеваний наследственно обусловленные формы составляют около 50%. Нарушение зрения входит в симптомокомплекс почти 280 наследственных заболеваний. Частота всех менделирующих глазных болезней составляет 5-7:1000. Эти нарушения обусловлены влиянием очень большого числа различных мутантных генов, обладающих широким плейотропным действием. Известно более 130 генов, нарушающих развитие и функционирование сетчатки (Гинтер Е.К., 2003).
Частота глазных заболеваний у детей в Российской Федерации составляет 16 случаев на 10 000 детей, слепоты и слабовидения - 1,6 случая на 10 000 детского населения (Нероев В.В., Хватова А.В., 2004). Доля наследственных заболеваний глаз составляет от 20% в развивающихся странах до 45% в странах с развитой экономикой и увеличивается в экономически развитых странах за счет снижения числа тяжелых инфекционных и других экзогенных заболеваний (Kocur Ivo, 2004).
Частота слепоты и слабовидения среди детей в Российской Федерации в 2004 г. составила 5,1 случая на 10 000. Распространенность наследственного поражения глаз в общей структуре глазных заболеваний составляет 30%. Среди причин слепоты доля генетически детерминированных нарушений органа зрения составляет 42-84% (Хлебникова О.В., 1998; Нероев В.В., Хватова А.В., 2004).
У 55% инвалидов в возрасте 19-50 лет заболевания глаз манифестировали в детском возрасте (Либман Е.С., 2000).
В структуре заболеваний органов зрения, приводящих к слепоте и слабовидению, растет удельный вес врожденных патологических изменений глаз. Обнаруживаемые нарушения неоднозначны по клиническим признакам. Иногда это небольшие аномалии, но нередко - грубые повреждения роговицы, хрусталика, сетчатки или зрительного нерва, приводящие к инвалидности (табл. 15-12).
Таблица 15-12. Патологические изменения глаз при наследственных болезнях
Наследственные заболевания характеризуются частым вовлечением в патологический процесс органа зрения. Среди всех органов и систем организма он занимает четвертое место после ЦНС, опорно-двигательного аппарата и сердечно-сосудистой системы по частоте поражения. Примерно одна четвертая часть зарегистрированных наследственных фенотипов характеризуется поражением глаз (Коста T. и др., 1985). Подобная высокая частота, очевидно, объясняется тем, что глаз как комплексный орган развивается из всех трех листков эмбриональной ткани и формируется под влиянием многих сотен генов. К настоящему времени картировано более 200 генных локусов, мутации в которых приводят к развитию моногенных заболеваний глаз, при которых отмечают либо изолированное поражение тканей глаза, либо сочетанное поражение с вовлечением других органов и систем организма (Freund C. et al., 1996). Сложные процессы нормального развития глаза и их многообразные нарушения затрудняют верификацию наследственных заболеваний, сопровождающихся поражением его отдельных элементов.
Трудности клинической диагностики наследственных синдромов, сопровождающихся поражением глаз, обусловлены как объективными, так и субъективными факторами. Среди первых можно выделить следующие:
-
многообразие наследственных синдромов, сопровождающихся патологическими изменениями органа зрения;
-
выраженный клинический полиморфизм офтальмологических нарушений;
-
генетическая гетерогенность изолированных и синдромальных форм нарушений;
-
различная степень экспрессивности и пенетрантности наследственных болезней, сопровождающихся поражением глаз;
-
малая изученность патогенетических механизмов развития наследственных синдромов, при которых в патологический процесс вовлечены элементы глаза;
-
недостаточное использование сложных методов исследований (биохимических, молекулярно-генетических, молекулярно-цитогенетических и др.) для верификации наследственной этиологии обнаруженных нарушений и др.
Определенные трудности создают и субъективные факторы, среди которых следует выделить мнение врачей о редкости наследственных болезней, существующие представления о безуспешности их лечения и отсутствие генетической настороженности врачей.
Наследственные болезни могут быть ассоциированы с любыми структурами глаза - роговицей, склерой, радужной оболочкой, хрусталиком, сосудистой оболочкой, сосудами, нейросетчаткой (включая палочки и колбочки) и зрительным нервом. Изменения могут быть изолированными или сочетаться с поражением одного или нескольких органов и систем организма.
Манифестация глазных нарушений при наследственных синдромах с поражением глаз и метаболических болезнях отличается значительной вариабельностью. Генетические дефекты глаз у детей чаще всего связаны с нарушением обмена веществ. При ряде наследственных болезней обмена глазные симптомы могут быть одними из признаков основного заболевания.
Несмотря на обширные исследования наследственных болезней обмена, патогенез офтальмологических нарушений остается недостаточно изученным.
Вовлечение глаз в патологический процесс в одних случаях может быть следствием прямого токсического действия метаболитов на орган зрения, в других - вызвано дефектами отдельных звеньев патогенеза или энергетической недостаточностью клеток, ведущей к нарушению метаболических процессов.
Обнаружение глазных нарушений может способствовать ранней диагностике наследственного заболевания, поскольку они могут быть его ведущим симптомом (Saudubray J.M., Charpentier C., 2001).
Наследственные болезни глаз обычно характеризуются двусторонним и симметричным поражением. Тяжелые нарушения зрения часто диагностируют на 2-м месяце жизни ребенка, когда начинает формироваться зрительный контакт между ним и окружающими людьми. В отдельных случаях аномалии глаз обнаружить относительно просто (например, выраженную катаракту при галактоземии). При других заболеваниях (например, при пероксисомных болезнях) нарушения обнаруживают только при исследовании глазного дна. У детей в неонатальном периоде оно может быть нормальным, в то время как отклонения диагностируют во время специальных исследований (например, при электроретинографии или регистрации вызванных зрительных потенциалов). Следует учитывать, что симптомы со стороны глаз могут манифестировать позднее.
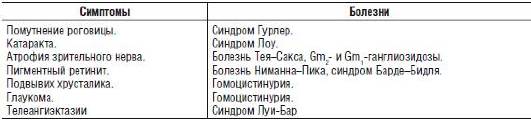
В табл. 15-13-15-17 представлены наследственные глазные болезни, характеризующиеся поражением отдельных структур глаза. Так, патологические изменения со стороны роговицы регистрируют при лизосомных болезнях, нарушениях липидного и аминокислотного обмена (см. табл. 15-13).
Таблица 15-13. Наследственные болезни обмена и дефекты роговицы
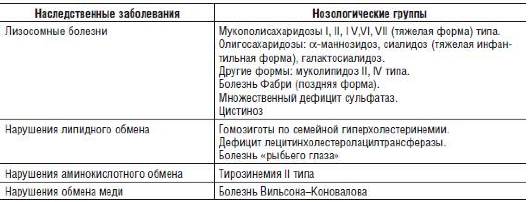
Множество наследственных дефектов метаболизма сопровождается поражением хрусталика. Среди них можно выделить основные виды нарушений обмена углеводов и аминокислот, лизосомные болезни, пероксисомные заболевания и др. (см. табл. 15-14).
Таблица 15-14. Дефекты хрусталика (катаракты) и наследственные болезни обмена
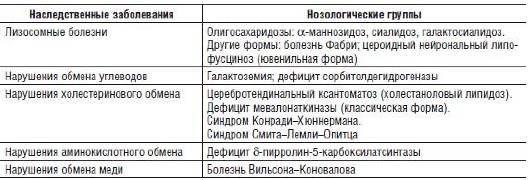
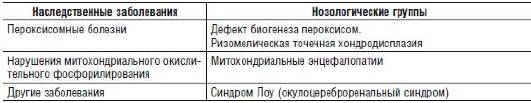
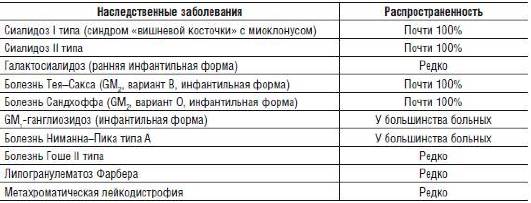
Обширная и генетически гетерогенная группа наследственных заболеваний сопровождается дегенерацией сетчатки (см. табл. 15-15). Описано более 400 наследственных нарушений, при которых в патологический процесс вовлекаются сетчатка, макула и сосудистая оболочка (Rattner et al., 1999). Известен 131 ген, мутации в котором специфически нарушают функцию сетчатки. Кроме того, идентифицировано более 20 генов, мутации в которых обусловливают дегенерацию фоторецепторов (Clarke G. et al., 2000). Функции многих белков, контролируемых этими генами, в полной мере остаются неисследованными. Некоторые белки являются структурными или транспортными, другие участвуют в фототрансдукции - многоэтапном процессе, подобном метаболическим цепям при наследственных болезнях обмена веществ. Процесс трансдукции считают геноконтролируемым, при этом мутации генов могут нарушать его и вызывать наследственные нарушения. Наследственные болезни сетчатки также могут быть связаны с мутациями генов, кодирующих белки фотоактивации.
Таблица 15-15. Наследственные дефекты обмена и дегенерация сетчатки
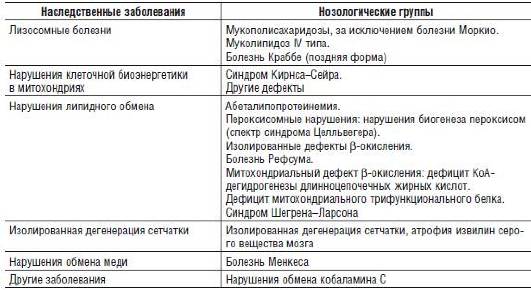
Среди генетических заболеваний детского возраста нередко обнаруживают наследственные нарушения, сопровождающиеся важными в диагностическом отношении офтальмологическими признаками. Среди них - феномен «вишневой косточки», имеющий важное дифференциально-диагностическое значение (см. табл. 15-16).
Разнообразные патологические изменения органа зрения в качестве клинических симптомов входят в фенотип многих наследственных болезней и дефектов. При этом обнаруживают поражение различных участков глаза: век, роговицы, радужной оболочки, хрусталика, сетчатки и зрительных нервов. Нередко отмечают сочетание глазных симптомов с умственной отсталостью. Подобные состояния могут быть обусловлены как хромосомными аномалиями, так и мутациями различных генов.
Таблица 15-16. Наследственные дефекты обмена, сопровождающиеся феноменом «вишневой косточки» на глазном дне
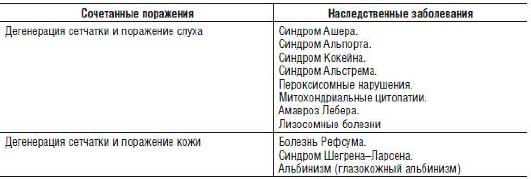
Наиболее распространенные наследственные болезни, сопровождающиеся снижением слуха и поражением кожи в сочетании с глазными нарушениями, представлены в табл. 15-17.
Таблица 15-17. Наследственные болезни, сопровождающиеся дегенерацией сетчатки, снижением слуха и (или) поражением кожи
Важное место в диагностике различных нейродегенеративных заболеваний принадлежит глазным симптомам, в частности оптической невропатии. Наиболее распространенные нозологические формы представлены в табл. 15-18.
Таблица 15-18. Оптическая невропатия как часть наследственных нейродегенеративных заболеваний

Клинический опыт диагностики наследственных болезней у детей показал, что она возможна при обращении особого внимания на сочетание поражения глаз и нервной системы, органа слуха, кожи, сердца, сосудов, печени, почек и скелета. Кроме того, необходимо учитывать существование дисморфических (грубых) черт лица, гормональных расстройств, нарушения поведения, прогрессирования заболевания и потерю ранее приобретенных навыков.
Следует помнить о выраженном клиническом полиморфизме и генетической гетерогенности изолированных форм поражения глаз, когда мутации в одном гене могут обусловливать разные по клинической картине заболевания и, напротив, мутации в разных генах приводят к развитию нарушений со сходными или идентичными клиническими признаками. Кроме того, аналогичные по клиническим симптомам поражения глаз могут быть обусловлены мутациями генов, не имеющих непосредственного отношения к развитию глаза, фоторецепции и другим специфическим офтальмологическим процессам.
Тщательный анализ родословной и знание диагностических признаков многообразных нозологических форм дает возможность определить этиологию патологических изменений и своевременно идентифицировать сочетанные поражения глаз и других органов и систем организма.
Нарушения слуха
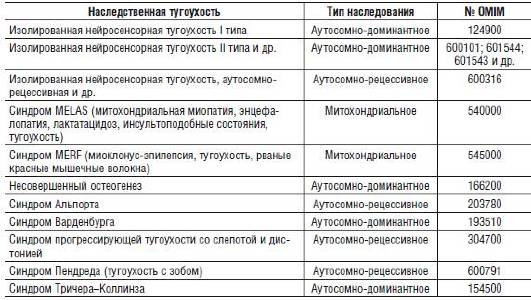
Нарушения слуха и тугоухость широко распространены у детей. По данным разных авторов, тугоухость у новорожденных обнаруживают с частотой 1:1000. Нарушения слуха входят в симптомокомплекс примерно 150 форм заболеваний. Известно, что примерно в 50% случаев происхождение глухоты у детей связано с генетическими факторами, при этом изолированные формы тугоухости составляют примерно 70%, а остальные 30% приходятся на синдромные формы. В подавляющем большинстве наследственные формы тугоухости (80%) наследуются по аутосомно-рецессивному типу, а около 20% - по аутосомно-доминантному. По 1% нарушений приходится на Х-сцепленное и митохондриальное наследование.
Аутосомно-рецессивные формы чаще врожденные и имеют более тяжелую степень выраженности (тугоухость III-IV степени). Аутосомно-доминантные формы манифестируют в период раннего детства и носят прогрессирующий характер. В настоящее время идентифицировано более 90 локусов изолированной тугоухости и более 40 генов, ассоциированных с тугоухостью, входящей в состав наследственных синдромов.
Наиболее распространенные формы поражения слуха представлены в табл. 15-19.
Таблица 15-19. Основные наследственные формы тугоухости и наследственные синдромы, сопровождающиеся ею
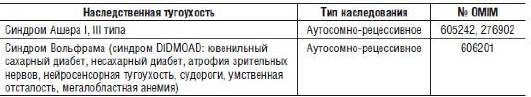
В медико-генетической практике наиболее часто синдромные формы тугоухости представлены синдромом Пендреда (нейросенсорная тугоухость с гипотиреоидным зобом), синдромом Джеруэлла-Нилсена (нейросенсорная тугоухость с аномалиями сердечной проводимости) и синдромом Ашера (нейросенсорная тугоухость с пигментным ретинитом).
Несмотря на то что глухота у детей и молодых людей во многих случаях обусловлена наследственными факторами, поставить точный диагноз не всегда легко. Его установление часто зависит от специального аудиологического обследования, поэтому во всех случаях ранней глухоты его считают обязательным.
В генетическом консультировании нуждаются две группы: родители ребенка, у которого обнаружено значительное снижение слуха, и молодые люди, страдающие глухотой и вступающие в брак с партнером, также страдающим ею. Правильному решению проблемы помогают следующие данные:
При рассмотрении генетической этиологии тугоухости всегда необходимо исключать нарушение, связанное с воздействием внешних факторов, и прежде всего стертую форму врожденной краснухи и цитомегаловирусную инфекцию.
Внедрение молекулярно-генетических технологий в анализ мутаций показало, что при ряде наследственных форм тугоухости обнаруживают интересный феномен: часть мутаций, обнаруженных в гене, манифестирует как доминантный признак, а часть - как рецессивный. Наследственная тугоухость отличается чрезвычайно высокой степенью гетерогенности. Именно поэтому при подозрении на ее наследственные формы рекомендовано по возможности проводить углубленный генетический анализ.
Морфогенетические варианты или микроаномалии развития: понятия, терминология и клиническое значение
Видимые отклонения в развитии органов и систем организма привлекают внимание врачей всех специальностей. Были предприняты неоднократные попытки использовать их в качестве теста для диагностики врожденных и наследственных заболеваний внутренних органов. Отклонения могут быть большими (с нарушением функций органов и систем или врожденными пороками развития) и малыми (без нарушения функций), чаще называемыми морфогенетическими вариантами развития или малыми аномалиями развития (МАР). Такие термины, как «стигмы дизэмбриогенеза », «диспластические болезни» и «дизонтогении», не совсем правильно отражают сущность МАР, и поэтому их не употребляют.
МАР, или врожденные морфогенетические варианты, выходят за пределы нормальных вариаций, но, в отличие от врожденного порока развития, не нарушают функцию органа. Они служат неспецифическими признаками эмбрионального дисморфогенеза и отражают небольшие отклонения гомеостаза, наследственные патологические изменения или нарушения, вызванные тератогенными факторами. Врожденные морфогенетические варианты обнаруживают и у здоровых людей, но существование нескольких признаков требует более внимательного обследования больного с целью диагностики врожденных или наследственных заболеваний.
Поскольку любое нарушение морфогенеза имеет диагностическую значимость, для обнаружения признаков дисморфогенеза необходимо внимательно осмотреть больного.
Ниже приведены иллюстрации наиболее распространенных признаков пре- и постнатального дисморфогенеза, оценка которых необходима для дифференциальной диагностики наследственных синдромов и болезней (рис. 15-1- 15-66). На каждом рисунке, как правило, можно видеть не один, а несколько признаков дисморфогенеза.

Рис. 15-1. Складчатая вялая кожа у девочки в возрасте шести лет: cutis laxa - кожа вялая
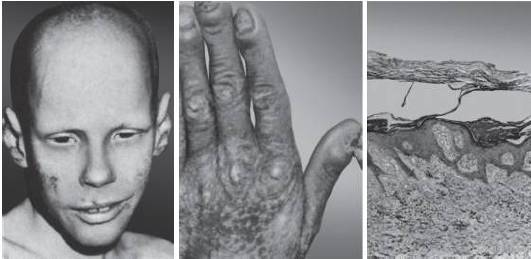
Рис. 15-2. Гиперкератоз (ихтиозоформная эритродермия)
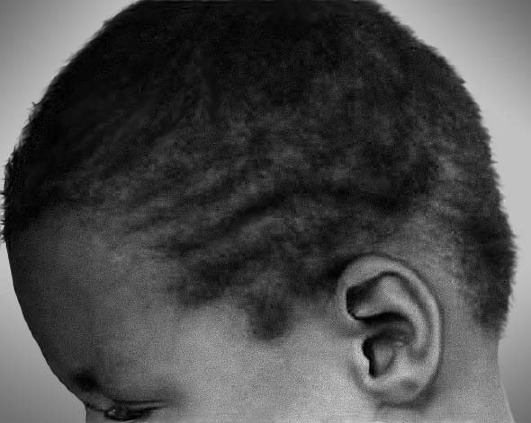
Рис. 15-3. Шерстистые волосы (синдром скрученных волос и глухоты)

Рис. 15-4. Седая прядь волос (синдром Варденбурга)
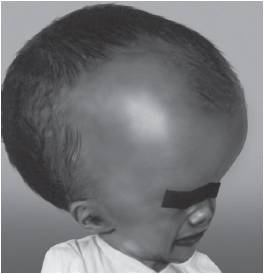
Рис. 15-5. Гидроцефалия (синдром Х-сцеплен ной гидроцефалии)
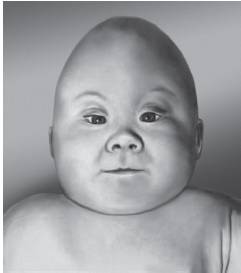
Рис. 15-6. Акроцефалополисиндактилия, или синдром Карпентера: акроцефалия, широкая переносица, низко расположенные уши, большие щеки, короткие глазные щели и короткая шея
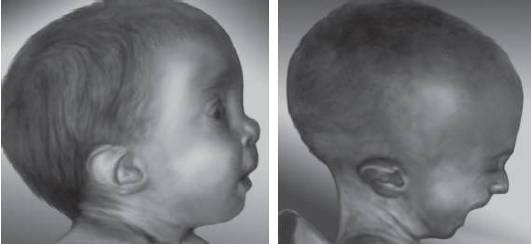
Рис. 15-7. Выступающий затылок, низко посаженные и отклоненные назад ушные раковины (трисомия 18)
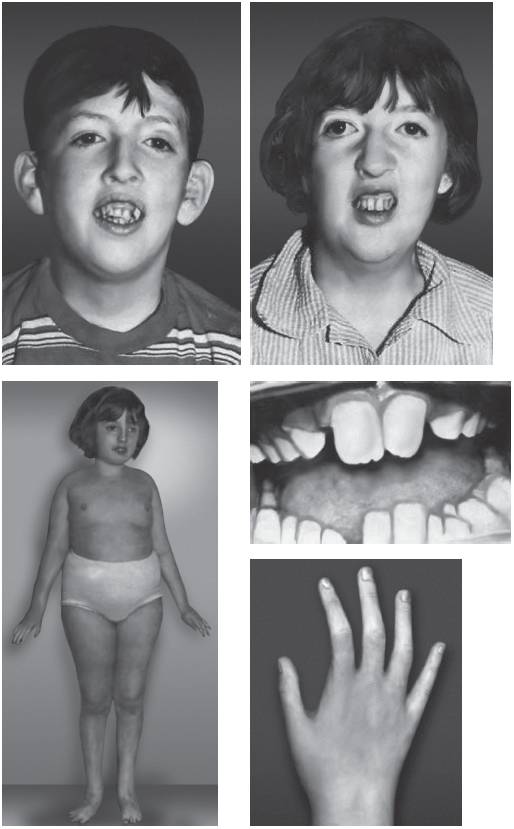
Рис. 15-8. Макротия и другие аномалии (синдром Коэна)
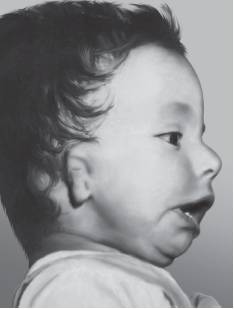
Рис. 15-9. Микротия, микрогнатия, макростомия (врожденная аномалия неясной этиологии)
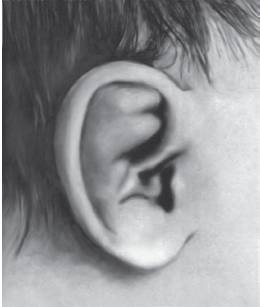
Рис. 15-10. Упрощенная форма завитка и утолщенный противозавиток (дистрофическая дисплазия)
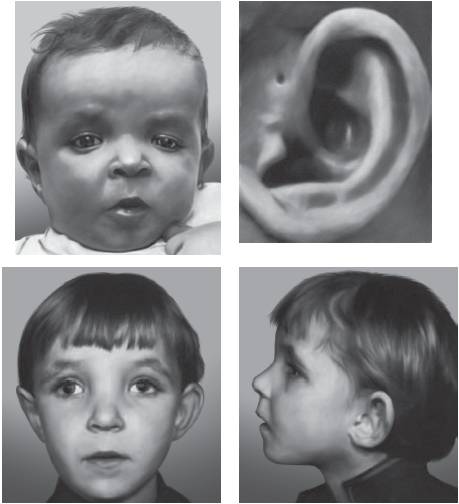
Рис. 15-11. Предушная фистула, отсутствие мочки уха, гипертелоризм (синдром «кошачьего глаза», или патологическое изменение хромосомы 22)
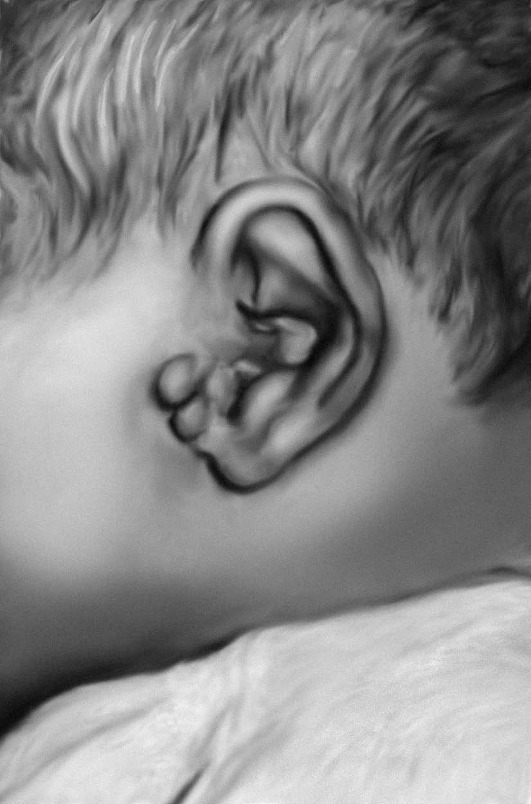
Рис. 15-12. Предушные папилломы (разные формы врожденных нарушений слуха)
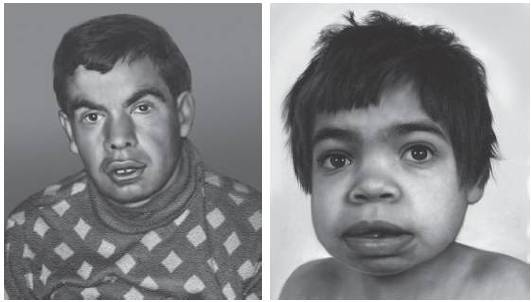
Рис. 15-13. Грубые черты лица (маннозидоз)
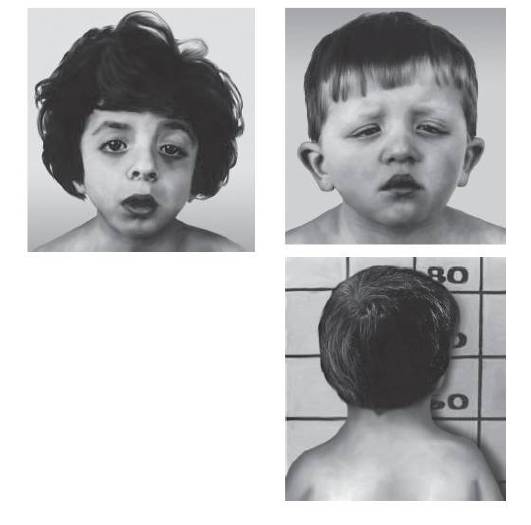
Рис. 15-14. Антимонголоидный разрез глаз, птоз, короткая шея (синдром Нунана)
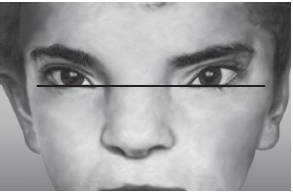
Рис. 15-15. Монголоидный разрез глаз (синдром Грейга)
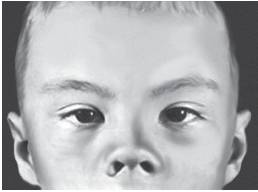
Рис. 15-16. Телекант, эпикант, плоская переносица и открытые вперед ноздри (синдром Элерса-Данло-Русакова)
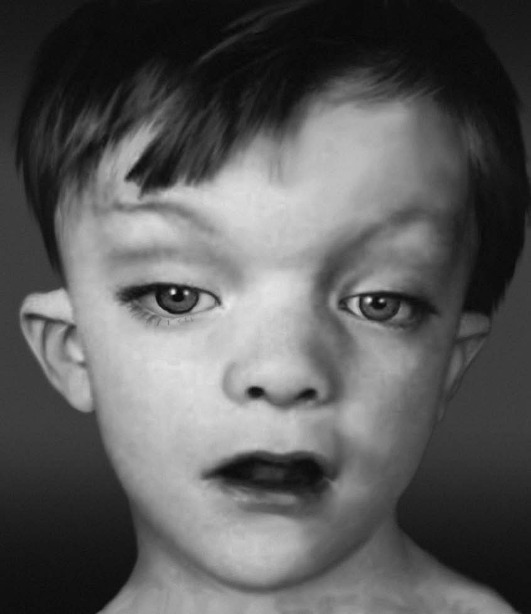
Рис. 15-17. Гипертелоризм, птоз и низко посаженные деформированные ушные раковины (синдром Аарскога)
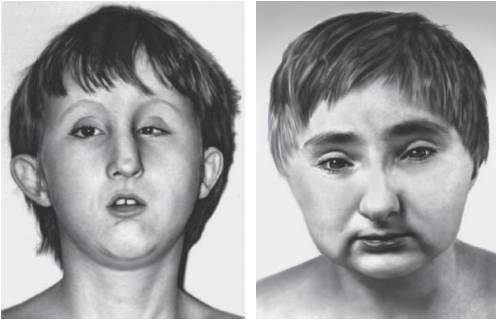
Рис. 15-18. Птоз, косоглазие, оттопыренные и низко посаженные ушные раковины с упрощенным рисунком, маленький рот (синдром Халлермана-Штрайфа)

Рис. 15-19. Косоглазие
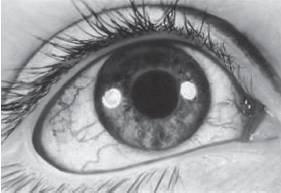
Рис. 15-20. Телеангиэктазии склеры (атаксия-телеангиэктазия)

Рис. 15-21. Синофриз (синдром Корнелии де Ланге)
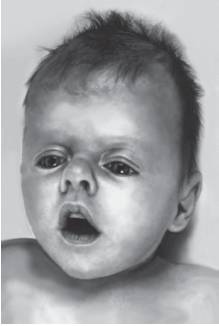
Рис. 15-22. Седловидная переносица, антимонголоидный разрез глаз (синдром Пфайфера)
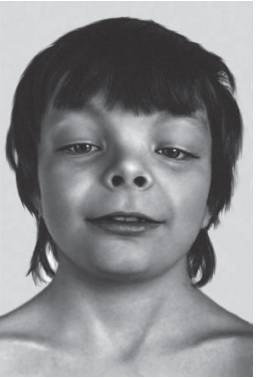
Рис. 15-23. Открытые вперед ноздри, короткий нос, антимонголоидный разрез глаз, гипертелоризм и широкая переносица (синдром Аарскога)
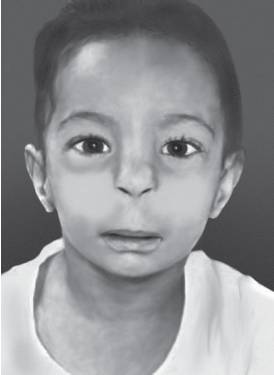
Рис. 15-24. Длинный фильтр, короткий нос с недоразвитыми крыльями, микрогения и низко посаженные уши (диабетическая эмбриопатия)

Рис. 15-25. Макрогения (синдром Х-сцепленной умственной отсталости)
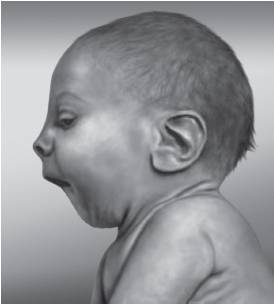
Рис. 15-26. Микрогения, крупные ушные раковины
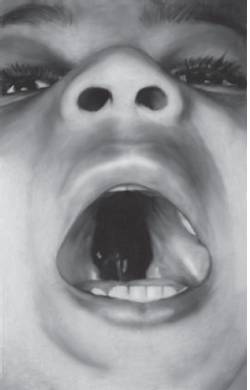
Рис. 15-27. Расщелина нёба (врожденный порок как следствие противосудорожного лечения беременной)
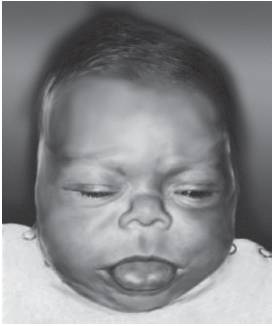
Рис. 15-28. Макроглоссия (синдром Беквита-Видеманна)
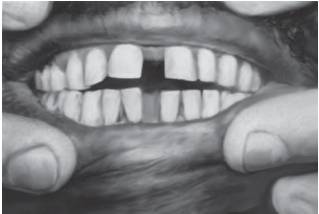
Рис. 15-29. Верхняя и нижняя диастема (распространена среди нубийцев Египта)
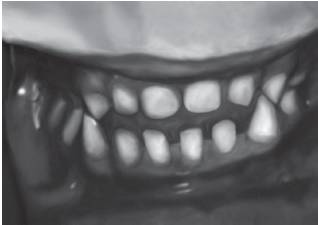
Рис. 15-30. Тремы - широкие промежутки между зубами (изолированный надклапанный стеноз аорты)
Рис. 15-31. Деформированная грудная клетка (килевидная или «куриная» грудь) при синдроме Марфана
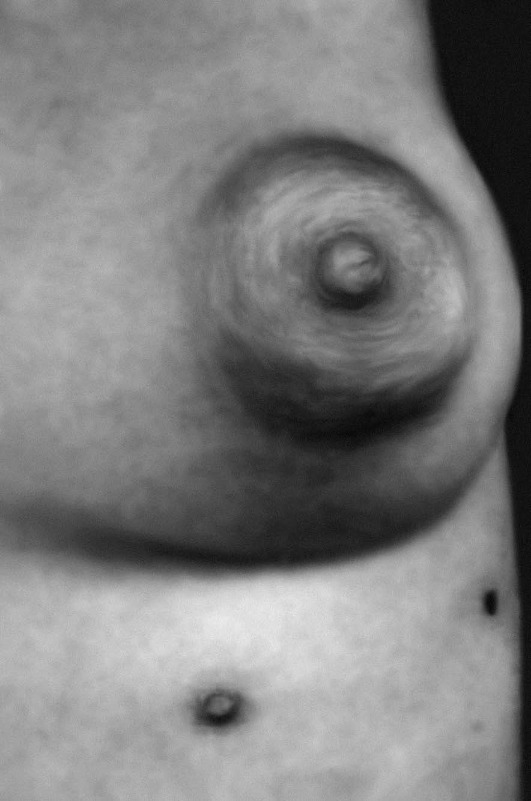
Рис. 15-32. Полителия (добавочный сосок), обнаруживаемая при частичной трисомии 12

Рис. 15-33. Пилонидальная ямка: обнаруживают при разных хромосомных и генных синдромах, а также при сахарном диабете у матери
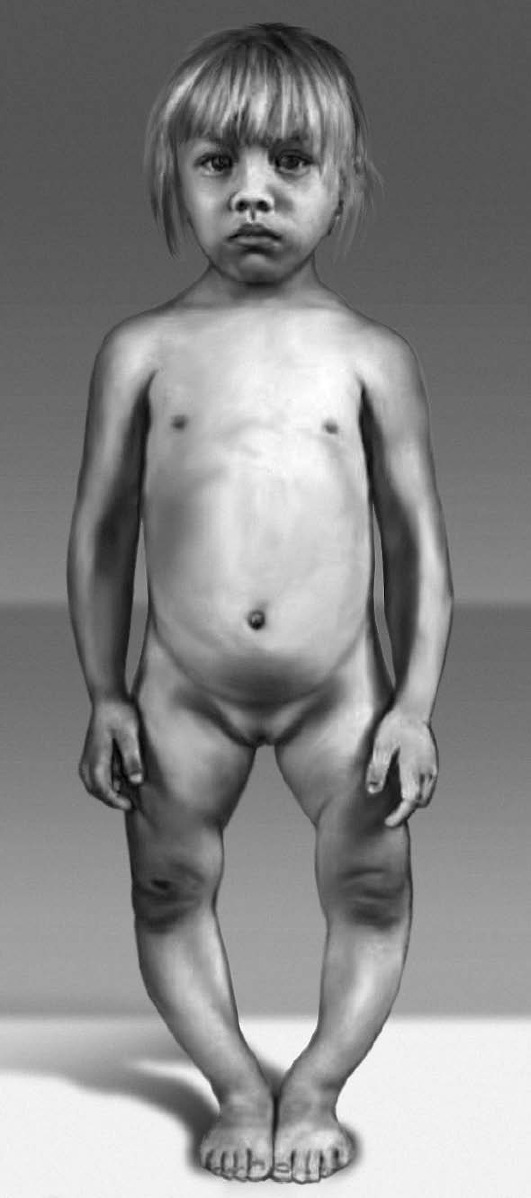
Рис. 15-34. Варусная деформация нижних конечностей и гипертелоризм сосков (гипофосфатемия или витамин D-резистентный рахит)
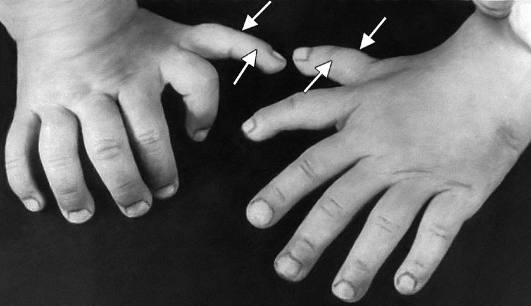
Рис. 15-35. Полидактилия преаксиальная
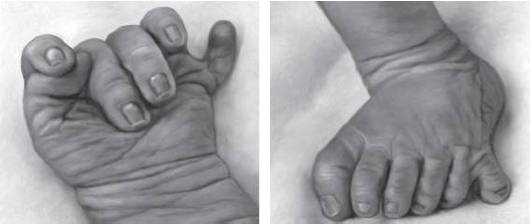
Рис. 15-36. Полидактилия кисти и стопы постаксиальная (синдром Смита-Лемли-Опитца)image::pic_0186.jpg[image]
Рис. 15-37. Олигодактилия кистей и стоп, гипоплазия отдельных пальцев и ногтей (постаксиальный акрофациальный дизостоз)
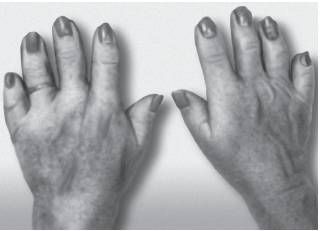
Рис. 15-38. Брахидактилия (тип А1)
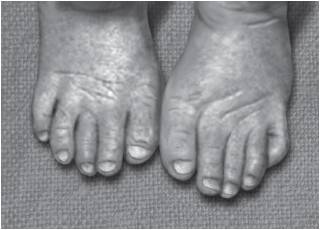
Рис. 15-39. Укороченные I пальцы стоп, гипоплазия ногтей: синдром Вольфа- Хиршхорна - делеция хромосомы 4р
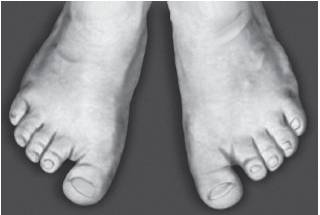
Рис. 15-40. Короткие пальцы стопы, короткие ногти, широкий I палец, сандалевидная щель (периферический дизостоз)
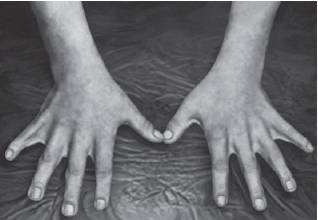
Рис. 15-41. Синдактилия кожная (синдром Аарскога)
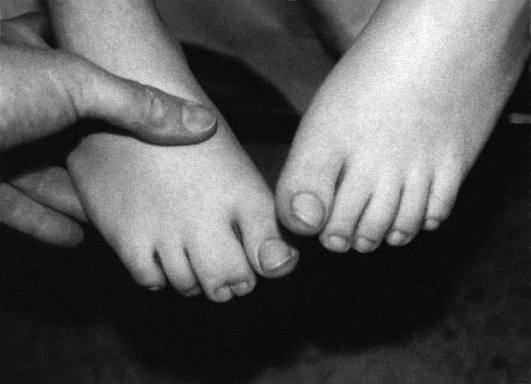
Рис. 15-42. Синдактилия II-III пальцев стоп разной выраженности (аминоптериновый синдром)
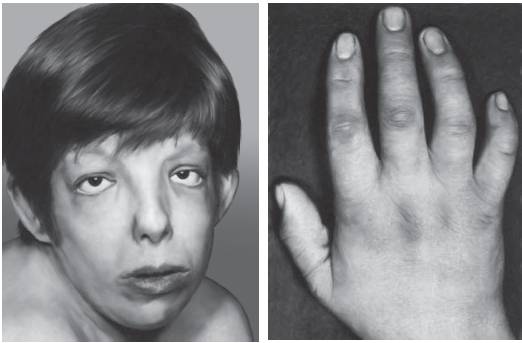
Рис. 15-43. Клинодактилия (синдром трисомии 9)
Рис. 15-44. Камптодактилия II пальца правой кисти и III пальца левой кисти (вальпроевый синдром)
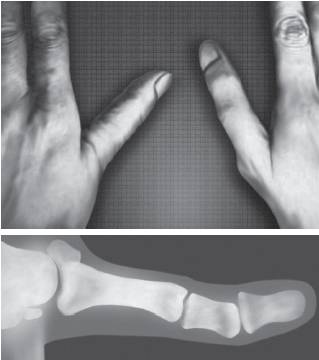
Рис. 15-45. Трехфаланговый I палец кисти (анемия и трехфаланговые I пальцы)
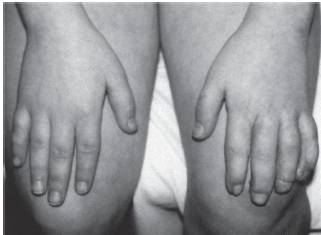
Рис. 15-46. Конусовидные пальцы (аминоптериновый синдром)

Рис. 15-47. Четырехпальцевая поперечная («обезьянья») ладонная складка: часто обнаруживают при хромосомных и генных болезнях, а также у 2-4% здоровых людей
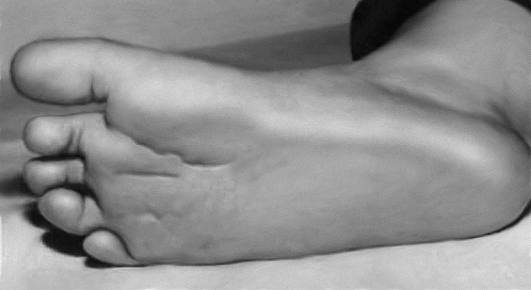
Рис. 15-48. Глубокая складка на стопе (трисомия 8, мозаичная форма)
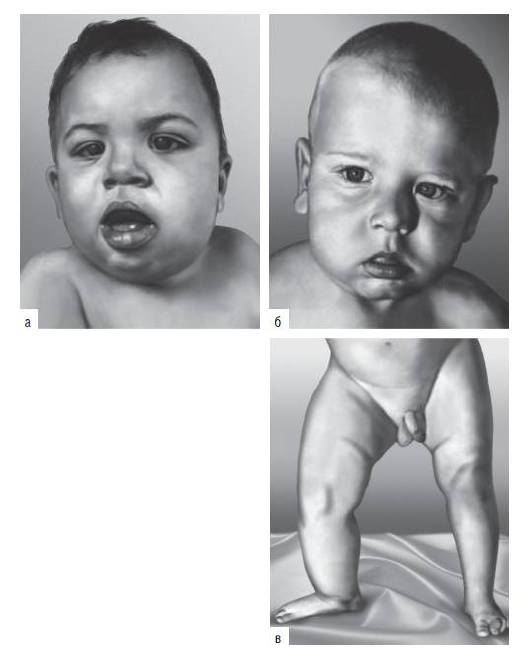
Рис. 15-49. Гемигипертрофия лица (а, б) и нижних конечностей (в) (гемигипертрофия)
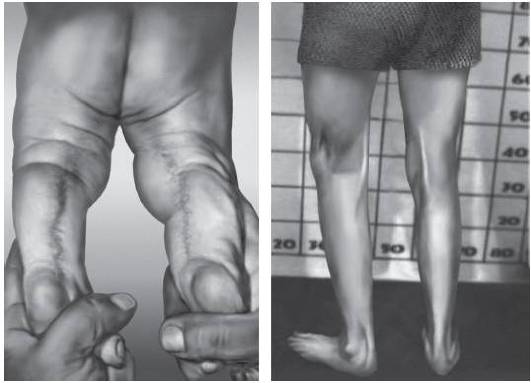
Рис. 15-50. Подколенная складка (синдром подколенного птеригиума)
Рис. 15-51. Гипоплазия концевых фаланг (брахидактилия, тип Д)
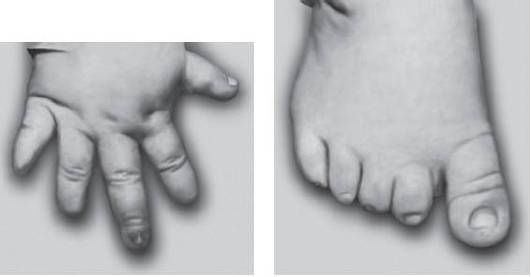
Рис. 15-52. Гипоплазия концевых фаланг и ногтей (гидантоиновый синдром)
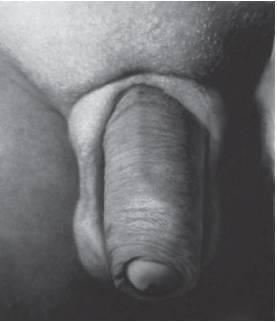
Рис. 15-53. Шалевидная мошонка (синдром Аарскога)
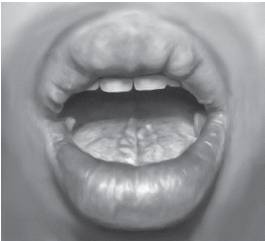
Рис. 15-54. Толстые губы (множественная эндокринная неоплазия - МЭН, тип II Б)
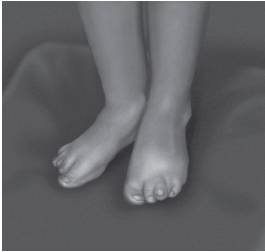
Рис. 15-55. Синдактилия
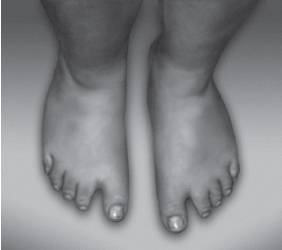
Рис. 15-56. Сандалевидная щель
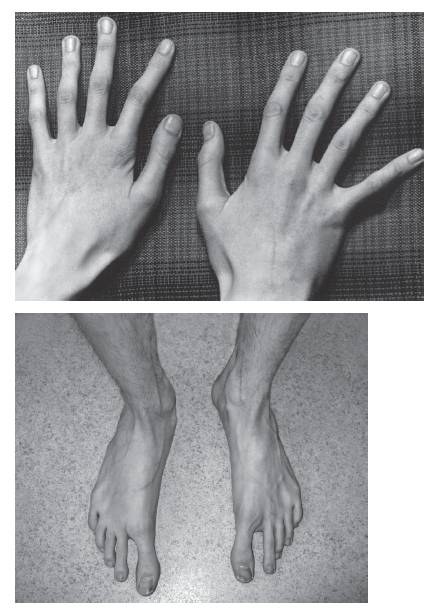
Рис. 15-57. Арахнодактилия (синдром Марфана)
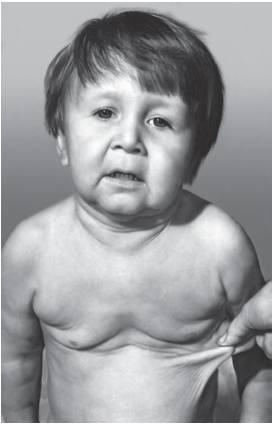
Рис. 15-58. Старческое лицо, «избыток» кожи
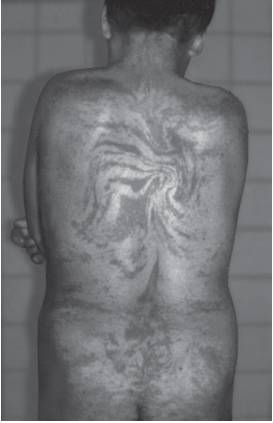
Рис. 15-59. Пигментация и депигментация в виде «брызг грязи» (синдром Блоха-Сульцбергера)
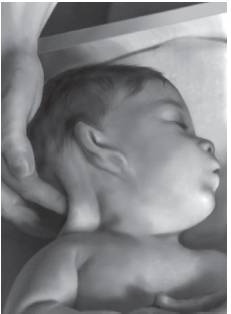
Рис. 15-60. Крыловидные складки кожи на шее (синдром Тернера)image::pic_0210.jpg[image]
Рис. 15-61. Гипертелоризм глаз, опущенные углы рта (слева)
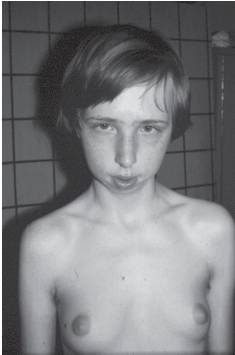
Рис. 15-62. Микрогения - недоразвитие нижней челюсти
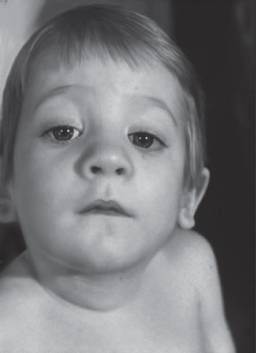
Рис. 15-63. Вывернутые ноздри, эпикант (синдром Шварца)image::pic_0213.jpg[image]
Рис. 15-64. Широкая переносица, короткий фильтр, телекант
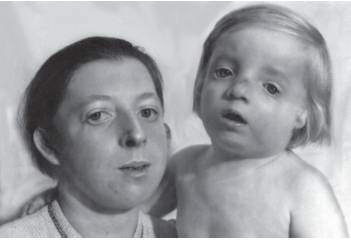
Рис. 15-65. Антимонголоидный разрез глаз, гипоплазия верхней челюсти, недоразвитие нижней челюсти, колобомы верхнего и нижнего века (синдром Франческетти)
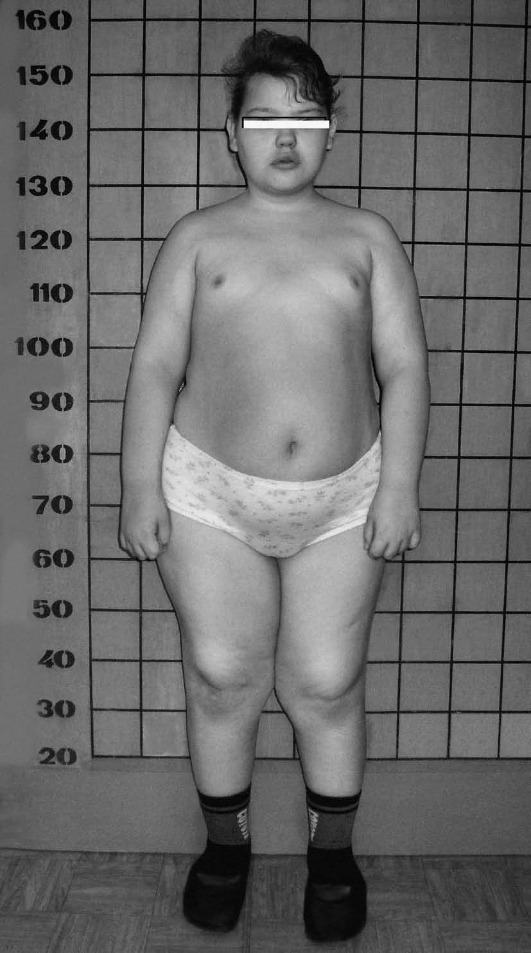
Рис. 15-66. Ожирение, гипогонадизм, умственная отсталость (синдром Бар-де-Бидля)
К сожалению, в настоящее время в нашей стране отсутствуют сведения о нормальной вариабельности микроаномалий в зависимости от генетической структуры и этнических особенностей популяций. Наличие таких данных повысило бы диагностические значения МАР.
Большое значение для диагностики наследственных болезней имеет анализ всех вариантов МАР. Например, при синдроме Вивера МАР можно считать большинство признаков: увеличенный бифронтальный размер черепа, плоский затылок, большие уши, гипертелоризм глаз, удлиненный фильтр, относительную микрогнатию и др. Следовательно, следует учитывать их популяционные и семейные частоты. Здесь еще раз подтверждается требование классической генетики о необходимости обследования не только пробандов, но и всех здоровых членов семьи. Его цель - определение зависимой или не зависимой от заболевания сегрегации МАР, при этом вероятность сочетания двух и более признаков, независимо встречающихся в популяции, выступить в качестве симптомов заболевания резко возрастает.
Таким образом, в диагностике наследственных заболеваний важнейшее значение приобретает тщательное (лучше - личное) обследование семьи.
При правильной регистрации врачи различных специальностей на первом этапе обследования детей с целью диагностики врожденных и наследственных заболеваний могут использовать МАР (с учетом возраста, национальности, семейного накопления) для клинического скрининга. Обнаружение четырех и более наиболее информативных МАР у ребенка позволяет отнести его к группе риска для последующего целенаправленного лабораторного (хромосомные болезни, генетические синдромы или болезни соединительной ткани) обследования.
У здоровых людей можно обнаружить врожденные отклонения морфогенеза, но существование нескольких признаков указывает на необходимость более внимательного обследования больного на предмет врожденных или наследственных патологических изменений.
Поскольку любое нарушение морфогенеза имеет диагностическое значение, при обследовании больных следует проводить внимательный осмотр с целью обнаружения признаков дисморфогенеза.
При анализе отдельных признаков наследственных болезней в первую очередь следует остановиться на значении МАР в формировании их клинической картины.
Всего различают более 80 различных МАР. В последние годы их стали использовать в диагностике врожденных и наследственных болезней.
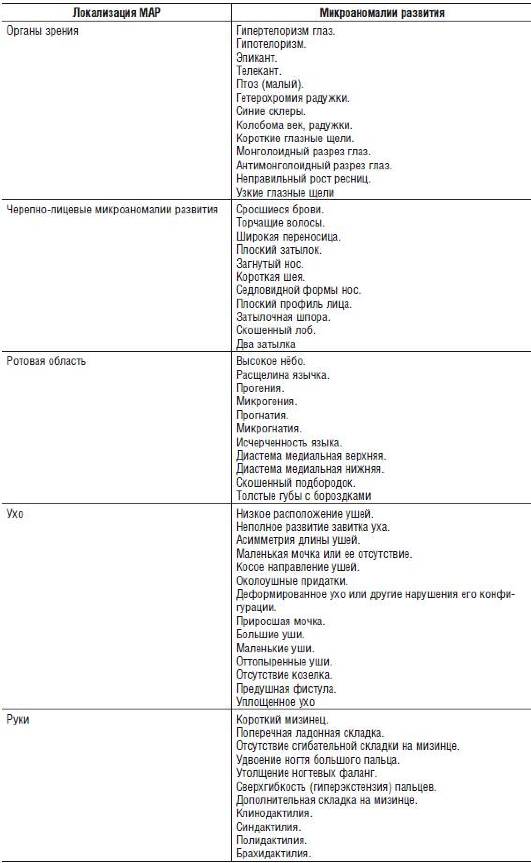
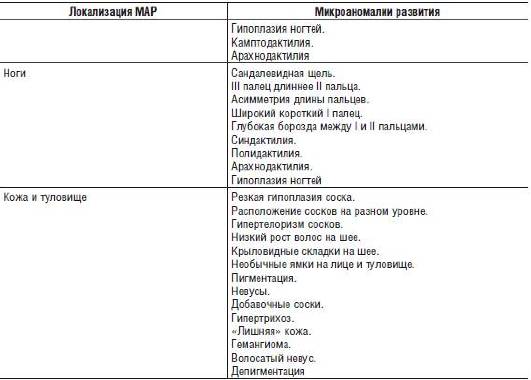
По месту локализации можно выделить МАР глаз, черепно-лицевые, ротовой области, уха, рук, ног, кожи и туловища (табл. 15-20).
Среди 86 наиболее распространенных МАР 17 можно характеризовать количественно, путем измерений или сопоставлений, а остальные - определить визуально.
Таблица 15-20. Основные микроаномалии развития
Ниже приведены 17 видов МАР, оцениваемых количественно, в том числе с помощью определенных индексов.
Плоский затылок в качестве МАР регистрируют, если у ребенка в положении лежа на спине затылок, шея и спина располагаются на одном уровне.
Гипертелоризм глаз оценивают по орбитальному индексу (ОИ). ОИ рассчитывают по формуле:
ОИ (%)=D/C,
где D - расстояние между внутренними углами глаз, С - окружность головы на уровне глаз. ОИ более 7,5% свидетельствует о гипертелоризме.
Для объективной оценки гипер- и гипотелоризма предлагают рассчитывать межорбитальный индекс (в %) путем деления расстояния между орбитами (см) на уровне внутренних углов глазных щелей на окружность головы (см) ×100. При гипертелоризме индекс составляет 6,8% и более.
Гипотелоризм глаз оценивают по ОИ по той же формуле, что и гипертелоризм. ОИ менее 3,8% свидетельствует о гипотелоризме.
Телекант - латеральное смещение внутренних углов глаз - рассматривают в качестве МАР в том случае, если воображаемая линия, проходящая через точку слезной железы, пересекает радужную оболочку. При количественной оценке телекант определяют по индексу, представленному отношением между внутренними углами глаз к расстоянию между зрачками. В норме он не превышает 0,6. Индекс более 0,6 свидетельствует о телеканте.
Монголоидный (азиатский) разрез глаз - косое расположение глазных щелей. Линейку располагают на переносице так, чтобы ее верхний край проходил через оба медиальных угла глаз. При монголоидном разрезе глаз латеральные углы расположены выше медиальных.
Антимонголоидный разрез глаз - косое расположение глазных щелей. Линейку располагают на переносице так, чтобы ее верхний край проходил через оба медиальных угла глаз. При антимонголоидном разрезе медиальные углы находятся выше, чем латеральные.
При косом расположении ушных раковин угол, образованный двумя линиями, одна из которых проходит вдоль ушной раковины, а вторая - вертикально через мочку уха, составляет более 20?.
Асимметрию ушных раковин определяют по различию размеров правого и левого уха, составляющему 15% и более.
Оттопыренные ушные раковины определяют путем измерения угла между задней поверхностью уха и мастоидальной областью. Если его величина приближается к 90?, это расценивают как МАР.
Низкое расположение ушных раковин оценивают путем сопоставления верхней точки прикрепления ушной раковины с уровнем латеральных углов глаз, при этом голова должна находиться в прямом положении
Короткий мизинец расценивают как МАР, если его верхний край совпадает или находится ниже середины II фаланги IV пальца.
Сандалевидную щель - широкое расстояние между I и II пальцем ноги - расценивают как МАР, если она оказывается равной или превышает ширину II пальца.
Синдактилию регистрируют как МАР при сращении пальцев, составляющем более 1/3 длины одного из пальцев.
Асимметрию длины пальцев определяют по сопоставлению длины всех пальцев.
Невусы (родинки) относят к МАР, если их диаметр равен 1 см и более.
Гипертелоризм сосков грудных желез определяют путем вычисления соскового индекса (%) - отношения расстояния между центрами сосков и окружностью грудной клетки на уровне них. Индекс более 28% расценивают как гипертелоризм, при этом положение плеч должно совпадать с горизонтальной линией.
Расположение сосков на разном уровне оценивают с помощью сантиметровой линейки, помещенной на грудной клетке, и последующего сопоставления с уровнем сосков. При измерении необходимо следить, чтобы положение плеч совпадало с горизонтальной линией.
МАР следует относить к клиническим тестам, которые в комплексе с другими методами можно использовать для диагностики хромосомных заболеваний, генетических синдромов, врожденных пороков развития, наследственных болезней соединительной ткани и др.
В то же время диагностическая значимость МАР невелика для больных с патологическими изменениями обмена аминокислот, углеводов, липидов, полигенными заболеваниями и наследственными тубулопатиями. В этом случае МАР не обнаруживают у 53-64% больных детей, а у остальных их число не превышает 1-3%, что аналогично особенностям их распределения у здоровых детей.
Как было сказано выше, оценку (диагностику) МАР можно использовать в качестве врачебного скрининга на долабораторном этапе обследования больных с врожденными и наследственными заболеваниями. В особую группу для последующего обследования необходимо выделять детей, у которых обнаруживают 4-5 МАР и более. Оценка МАР с учетом характера и количества ускоряет процесс обследования и повышает его точность.
Ряд сочетаний МАР, с одной стороны, повышает их диагностическую значимость, а с другой - придает определенную специфичность. Например, сочетание высокого нёба, сандалевидной щели и арахнодактилии позволяет предположить синдром Марфана; утолщение ногтевых фаланг, высокое нёбо и низкое расположение ушных раковин - синдром Рубинштейна-Тейби; скошенный лоб и гипертелоризм глаз - синдром Крузона; плоский профиль лица, телекант и гетерохромия радужек - синдром Варденбурга; антимонголоидный разрез глаз и микрогения - синдром Франческетти и др.
СПИСОК РЕКОМЕНДУЕМОЙ ЛИТЕРАТУРЫ
Акуленко Л.В., Богомазов Е.А., Захарова О.М. и др. Медицинская клиническая генетика для стоматологов. - М.: ГЭОТАР-Медиа, 2008. - 398 с.
Аномалии развития (иллюстрированное пособие для врачей) / Под общ. ред. В.В. Красильникова. - СПб.: ФОЛИАНТ, 2007. - 336 с.
Баранов В.С., Кузнецова Т.В. Цитогенетика эмбрионального развития человека: научнопрактические аспекты. - СПб., 2007. - 640 с.
Беляков ЮА. Наследственные болезни и синдромы в стоматологической практике. - М.: Медицина, 2008. - 238 с.
Бочков Н.П., Пузырев В.П., Смирнихина C.A. Клиническая генетика. - М.: ГЭОТАРМедиа, 2011. - 544 с.
Гинтер Е.К. Медицинская генетика. - М.: Медицина, 2003. - 448 с.
Демикова Н.С. Врожденные пороки развития. Руководство по педиатрии. - М.: Династия, 2007. - 544 с.
Иллариошкин С.Н., Иванова-Смоленская И.А., Маркова Е.Д. ДНК-диагностика и медикогенетическое консультирование в неврологии. - М.: Медицинское информационное агентство, 2002. - 591 с.
Кадурина Т.И., Горбунова В.Н. Дисплазия соединительной ткани. Руководство для врачей. - СПб.: Элби, 2009. - 704 с.
Краснопольская К.Д., Евдокименкова В.Н., Захарова Е.Ю. и др. Новые подходы к диагностике и профилактике наследственных болезней обмена // Принципы организации и методические основы профилактики инвалидизирующих наследственных болезней: Сборник. - М., 1996.- С. 35-37.
Лазюк Г.И. Тератология человека. - М.: Медицина, 1991. - 480 с.
Лазюк Г.И., Лурье И.В., Черствой Е.Д. Наследственные синдромы множественных пороков развития. - М., 1983. - 204 с.
Мордовцев В.Н., Мордовцева В.В. Наследственные болезни и пороки развития кожи. Клиника. Морфология. Лечение. Атлас. - М.: Наука, 2004. - 174 с.
Назаренко С.А. Наследственные болезни, детерминированные однородительскими ди сомиями, и их молекулярная диагностика. Молекулярно-биологические технологии в медицинской практике. - Новосибирск: Альта-Виста, 2003. - С. 75-91.
Нероев В.В., Хватова А.В. Основные направления «Российской целевой программы по ликвидации устранимой детской слепоты» // Материалы 2-го Российского межрегионального симпозиума «Ликвидация устранимой слепоты: всемирная инициатива ВОЗ. Ликвидация детской слепоты». - М., 2004. - С. 39-50.
Наследственные болезни нервной системы: Руководство для врачей / Под общ. ред. Ю.Е. Вельтищева. - М.: Медицина, 1998. - 496 с.
Новиков П.В. Семиотика наследственных болезней у детей (симптом, синдром, болезнь). - М.: Триада-Х, 2009. - 432 с.
Пренатальная диагностика наследственных и врожденных болезней / Под общ. ред. Э.К. Айламазяна. - М.: МЕДпресс-информ, 2006. - 416 с.
Светлов П.Г. Теория критических периодов развития и ее значение для понимания принципов действия среды на онтогенез // Вопросы цитологии и общей физиологии» / Под общ. ред. В.Я. Александрова. - М., 1960. - С. 263.
Falkner P., Tanner J.M. Human Growth. - New York: Plenum Press, 1986. - P. 1-12.
Fiengold M., Pashayan H. Genetics and Birth defects in Clinical Practice. - Boston; Toronto: Brown and Company, 1983. - 245 p.
Marden P.M., Smith D.W., McDonald M.S. Congenital anomalies in the newborn infants, including minor variations // J. Pediatr. - 1964. - Vol. 64, N 3. - P. 357-371.
Mehes K. Minor malformations in the neonate. - Stalder; Budapest: Akad kiado, 1983. - 129 p.
Rattner A., Sun H., Nathans J.U. Molecular genetics of human retinal disease // Annu. Rev. Genet. - 1999. - Vol. 33. - P. 89-131.
Saudubray J.M., Charpentier C. Clinical phenotypes: diagnosis/algorithms. The Metabolic and Molecular Bases of Inherited Disease. - New York: McGraw-Hill, 2001. - P. 1327-1403.
Scriver C.R., Beаudet A.L, Sly W.S., Valle D. The Molecular Basis of Inherited Metabolic Diseases. - New York: McGraw-Hill, 2001. - Vol. 1-4.
Smith D.W. Smith?s Recognizable Patterns of Malformation. - Philadelphia: W.B. Saunders, 1997. - 704 p.