Детские болезни : учебник / Геппе Н. А. - Москва : ГЭОТАР-Медиа, 2018. - 760 с. - ISBN 978-5-9704-4470-2 |
8.2. Гломерулонефриты
Гломерулонефриты (ГН) — неоднородная группа приобретенных заболеваний почек, различных по этиологии, клинико-морфологическим проявлениям, течению и исходу, преимущественно носящих характер иммунного воспаления с первичным поражением клубочков и вторичным вовлечением в патологический процесс канальцев и интерстиция.
Эти заболевания — одна из ведущих причин формирования хронической болезни почек, под которой понимают наличие любых клинических и лабораторных маркеров повреждения почек, персистирующих более 3 мес, вне зависимости от нозологического диагноза.
Классификация
ГН может быть:
По течению выделяют:
Ведущей является морфологическая классификация, основанная на результатах исследования почечного биоптата. ГН, при которых выявляют морфологические признаки воспаления, определяемые по наличию гиперклеточности клубочка, обусловленной пролиферацией собственных (резидентных) клеток и лейкоцитарной инфильтрацией (нейтрофилами, моноцитами, реже лимфоцитами), считают пролиферативными. Они могут быть эндокапиллярными, когда увеличено количество эндотелиальных и мезангиальных клеток, и экстракапиллярными, когда увеличено число париетальных эпителиальных клеток. Эти заболевания чаще всего проявляются нефритическим синдромом. К ним относят:
Для непролиферативных ГН характерно поражение слоев клубочкового фильтра (подоцитов и базальной мембраны), служащих основным барьером для белков. К ним относят:
Эти заболевания проявляются нефротическим синдромом.
Однако однозначной связи морфологических вариантов с этиологией, клинической картиной и патогенезом нет. Многие из них имеют несколько этиологических факторов, иммунных механизмов и клинических проявлений.
Этиология
Этиология большинства форм ГН неизвестна.
Ее чаще можно установить при остром процессе (в 80–90% случаев) и очень редко (в 5–10%) при хроническом. Среди этиологических факторов имеют значение стрептококковая, стафилококковая и другие бактериальные инфекции. Доказана роль вируса ВГВ и HCV, вирусов герпеса (1-го и 2-го типов, цитомегаловируса), ВИЧ, энтеровирусов. Возможно развитие ГН на фоне паразитарных заболеваний (например, при малярия); токсического воздействия лекарств (препаратов золота, пеницилламина, НПВП и др.), у подростков — алкоголя, наркотиков. Иногда начало заболевания бывает спровоцировано неинфекционными факторами (профилактическими прививками, введением сыворотки, охлаждением и т.д.), вызывающими аллергическую реакцию или служащими «пусковым» моментом на фоне предшествующей сенсибилизации организма.
Патогенез
Инфекционные или иные стимулы вызывают иммунный ответ, представленный иммунокомплексным или антительным механизмом.
-
Иммунокомплексный механизм заключается в образовании иммунных комплексов, состоящих из Аг и синтезированных против них АТ, которые могут формироваться в крови больного (циркулирующие иммунные комплексы) и затем фиксироваться на базальной мембране клубочков и в мезангии или непосредственно в гломерулярной ткани (in situ). Образованные иммунные комплексы классическим или альтернативным путем активируют систему комплемента, вследствие чего происходит выброс факторов, обеспечивающих хемотаксис и адгезию полиморфноядерных лейкоцитов и тромбоцитов, дегрануляцию базофилов и тучных клеток, формирующих мембраноатакующий комплекс (С5b–C9), непосредственно повреждающий гломерулярную базальную мембрану. Это приводит к активации тромбоцитов, свертывающей и калликреин-кининовой систем крови.
-
Антительный механизм обусловлен фиксацией цитотоксических АТ, направленных против собственных Аг в тканях клубочка с последующей активацией комплемента и распространением иммуновоспалительного повреждения. «Классический» Аг при антительном ГН — гликопротеин ГБМ. Наряду с этим цитотоксические АТ могут связываться с Аг подоцитов и мезангиальных клеток.
-
В клубочках накапливается множество клеток (нейтрофилов, эозинофилов, моноцитов-макрофагов, тромбоцитов), продуцирующих в большом количество различные медиаторы воспаления: цитокины (ФНОα, ИЛ-1, ИЛ-6, интерферон γ), факторы роста (тромбоцитарный фактор роста, трансформирующий фактор роста β), протеолитические ферменты, активные радикалы кислорода, липидные медиаторные субстанции, провоспалительные простагландины, вазоактивные субстанции. Цитокины и факторы роста вырабатываются как инфильтрирующими воспалительными клетками так и собственными клетками клубочков и интерстиция.
-
Одновременно с пролиферацией клеток клубочков (мезангиальных, эндотелиальных или эпителиальных) усиливается синтез (экспансия) внеклеточного матрикса. При длительном воспалении развиваются гломерулосклероз и интерстициальный фиброз — морфологическая основа хронической почечной недостаточности.
-
В прогрессировании почечного повреждения важную роль играют неиммунные механизмы:
-
гемодинамические факторы — внутриклубочковая гипертензия и гиперфильтрация, связанные с повышением системного АД и гиперфункцией оставшихся нефронов; они усиливают проницаемость гломерулярного фильтра, что способствует отложению различных макромолекул плазмы в тканях нефрона, ведут к активации ренин-ангиотензин-альдостероновой системы, повышению синтеза ангиотензина II — важного фактора пролиферации клеток почечных клубочков;
-
выраженная, длительная протеинурия действует как «внутренний токсин», поскольку реабсорбция профильтровавших белков активирует эпителий проксимальных канальцев, что стимулирует высвобождение им воспалительных и вазоактивных веществ — хемокинов и эндотелина; последние, синтезируясь в большом количестве, привлекают другие клетки, вызывающие воспалительную интерстициальную реакцию, предшествующую развитию тубулоинтерстициального фиброза;
-
гиперлипидемия оказывает повреждающее действие на эндотелий капилляров клубочков, а продукты перекисного окисления липидов стимулируют пролиферацию мезангия и синтез коллагена (развитие гломерулосклероза).
-
Воздействие на патогенетические иммунные процессы и неиммунные факторы прогрессирования изменений в почках — основной принцип лечения ГН, заключающийся:
8.2.1. Острый постстрептококковый гломерулонефрит
Самый частый вариант ГН в детском возрасте. Ежегодно в мире выявляют 470 тыс. новых случаев острого постстрептококкового ГН, из них 400 тыс. — у детей. Наиболее высокая заболеваемость в возрасте от 5 до 12 лет, мальчики болеют чаще девочек. Обычно наблюдают спорадические случаи заболевания, но возможны и эпидемические вспышки.
Этиология
Этиология — БГСА.
В основном нефритогенные М-штаммы: 1, 2, 4, 12, 49, 55 57, 60. Это подтверждают бактериологическое исследование (выделение чистой культуры стрептококка из зева, из очагов пиодермии) и высокие уровни АТ к стрептококку (антистрептолизина О, анти-ДНКазы В, АТ к М-протеину и др.). Предполагают, что нефротропными Аг являются нефрит-ассоциированный рецептор плазминового комплекса, стрептококковый пирогенный экзотоксин В и его предшественник зимоген. Возможно развитие острого постстрептококкового ГН, вызванного β-гемолитическим стрептококком группы С.
Заболевание развивается через 1–3 нед после фарингеальной (ангина, фарингит, скарлатина) или через 3–6 нед после кожной инфекции (пиодермии). Возможно развитие острого постстрептококкового ГН после отита, лимфаденита, остеомиелита.
Патогенез
Патогенез: иммунокомплексное заболевание, механизмом развития которого может быть:
-
первичная фиксация Аг нефритогенных штаммов стрептококков в клубочках почек (в гломерулярной базальной мембране и/или мезангиуме), связывание их с аутоантителами с образованием иммунных комплексов in situ и активацией комплемента (данный иммунный процесс наиболее вероятен);
-
отложение в гломерулах циркулирующих иммунных комплексов, в состав которых входит стрептококковый Аг;
-
фиксация стрептококкового Аг в почечной ткани с развитием феномена молекулярной мимикрии с перекрестным взаимодействием АТ с гломерулярными структурами.
Морфология
Морфология: диффузный пролиферативный эндокапиллярный ГН с лейкоцитарной (преимущественно нейтрофильной) инфильтрацией капиллярных петель (при световой микроскопии, рис. 8.4).
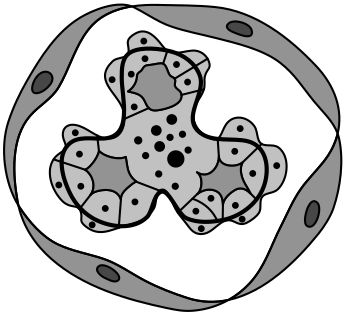
При иммунофлюоресцентном исследовании обнаруживают диффузное отложение IgG и C3-компонента комплемента гранулярного характера в мезангии и капиллярной стенке. Патогномоничный признак острого постстрептококкового ГН при электронной микроскопии — субэпителиальные плотные куполообразные депозиты в виде горбов. Возможно (в редких случаях) формирование полулуний, определяющее неблагоприятный прогноз заболевания.
Клиническая картина
Клиника: остронефритический синдром или бессимптомная гематурия.
Заболевание дебютирует внезапно после перенесенной стрептококковой инфекции. Возникают симптомы интоксикации (недомогание, вялость, плохой аппетит, тошнота, бледность, субфебрильное повышение температуры тела), могут быть боли в поясничной области, причина которых — растяжение капсулы почек вследствие отека их паренхимы.
При типичном циклическом течении острого постстрептококкового ГН развивается остронефритический синдром, который включает экстраренальные и ренальные симптомы.
Экстраренальные (клинические) симптомы:
-
отеки появляются у большинства пациентов, преимущественно на лице (более заметные по утрам), иногда — распространенные с развитием асцита и гидроторакса, что чаще наблюдается у детей младшего возраста; причина отеков — гиперволемия вследствие снижения фильтрации и задержки натрия;
-
макрогематурия (в 50% случаев) — моча становится темно-коричневой (что связано с изменением цвета гемоглобина в кислой среде), реже приобретает красный цвет и имеет вид «мясных помоев» (при щелочной реакции мочи);
-
АГ (у 75% пациентов) — повышено как систолическое, так и диастолическое АД. АГ обусловлена увеличением объема циркулирующей крови из-за задержки натрия и жидкости, а также повышением сердечного выброса и периферического сопротивления сосудов.
Ренальные (лабораторные) симптомы:
В начале заболевания возможна абактериальная лейкоцитурия (в основном лимфоцитурия), отражающая острый иммуновоспалительный процесс в клубочках. В редких случаях (у 2–4% пациентов) развивается нефротический синдром.
Лабораторная данные:
-
повышение уровня АТ к Аг стрептококка (антистрептолизина О, анти-ДНКазы В и др.); пик антистрептолизина О достигается через 2–4 нед после фарингита, уровень его остается повышенным в течение нескольких месяцев;
-
гипокомплементемия — снижение активности С3 и общей гемолитической активности комплемента наблюдают почти у всех в течение 4–8 нед;
-
умеренный лейкоцитоз (вторичный по отношению к перенесенной стрептококковой инфекции), иногда повышение СОЭ, снижение уровней гемоглобина и тромбоцитов в результате гемодилюции.
В типичных случаях острый постстрептококковый ГН имеет циклическое течение — вслед за остронефритическим периодом происходит обратное развитие симптомов, затем наступает клинико-лабораторная ремиссия. Отеки и АГ обычно купируются быстро (в течение 10–14 дней), изменения в моче (протеинурия, микрогематурия) исчезают через несколько месяцев (1,5–2, иногда позже).
В настоящее время заболевание чаще имеет ациклическое течение с изолированным мочевым синдромом (микрогематурия, цилиндрурия, протеинурия менее 1 г/сут). Экстраренальные проявления отсутствуют либо выражены столь незначительно и кратковременно, что остаются незамеченными.
При тяжелом течении острого ГН в начальном период возможны угрожающие жизни осложнения:
-
гипертензионная энцефалопатия — эклампсия, обусловленная спазмом сосудов головного мозга и его отеком (головная боль, рвота, снижение зрения, возможны тонико-клонические судороги, потеря сознания); при отсутствии своевременной адекватной терапии возможен летальный исход от кровоизлияния в головной мозг;
-
острое повреждение почек, проявляющееся резким сокращением или прекращением мочевыделения (анурия), снижением скорости клубочковой фильтрации, гипергидратацией, азотемией (повышением уровней мочевины и креатинина сыворотки), гиперкалиемией, метаболическим ацидозом;
-
острая сердечно-сосудистая недостаточность — у детей это осложнение возникает редко.
Биопсия почки целесообразна при долго сохраняющихся экстраренальных симптомах, изменениях в моче, сочетании нефритического синдрома с нефротическим, длительной депрессии С3, отсутствии восстановления скорости клубочковой фильтрации и уровня азотистых шлаков через 2–3 нед от дебюта болезни. Последнее требует исключения быстропрогрессирующего ГН, отсроченная терапия которого приводит к быстрому и необратимому нарушению почечных функций.
Острый постстрептококковый ГН следует дифференцировать от других пролиферативных вариантов ГН, проявляющихся остронефритическим синдромом (экстракапиллярного, мембранопролиферативного) или изолированной гематурией (IgA-нефропатии), наследственных гломерулопатий (болезнь тонких базальных мембран, синдром Альпорта), мочекаменной болезни.
Лечение
Лечение больных острым постстрептококковым ГН проводят в стационаре.
-
В остром периоде — постельный режим до купирования экстраренальных симптомов (исчезновения отеков, снижения АД).
-
Ограничение приема жидкости, натрия хлорида, белка. Объем жидкости рассчитывают по диурезу предыдущего дня, учитывая потери на перспирацию. В первые дни болезни при олигурии, АГ, распространенных отеках, снижении клубочковой фильтрации назначают бессолевой стол (после купирования этих симптомов соль постепенно добавляют — 0,5– 1 г/сут), ограничивают белок до 1,0–0,5 г/кг в сут, продукты, богатые калием (в том числе фруктовые и овощные соки). Общая энергетическая ценность пищи должна соответствовать потребностям ребенка в основном за счет углеводов и жиров.
-
Антибактериальная терапия показана при сохранении активности стрептококковой инфекции к моменту диагностики ГН, положительных результатах бактериологического исследования (мазка из зева или посева из очагов пиодермии) на БГСА. Используют препараты с низкой нефротоксичностью, дозу определяют с учетом скорости клубочковой фильтрации. Препараты первого выбора — антибиотики пенициллинового ряда (амоксициллин + клавулановая кислота, ампициллин и др.), курс 7–10 дней. Альтернативные препараты (второго выбора): макролиды 2-го и 3-го поколений (азитромицин, кларитромицин), цефалоспорины 2-го поколения (цефуроксим и др.).
-
Диуретическую терапию назначают при выраженных отеках, АГ: петлевые диуретики — фуросемид (1,5–2 мг/кг в сут парентерально 1–2 дня, затем внутрь 3–5 дней). При скорости клубочковой фильтрации выше 30 мл/мин можно использовать тиазидные препараты. Применение калийсберегающих диуретиков ограничено риском развития гиперкалиемии.
-
Гипотензивные средства необходимы при выраженной АГ, когда диуретической терапии недостаточно для контроля АД. Используют блокаторы медленных кальциевых каналов (нифедипин 0,25–0,5 мг/кг в сут, амлодипин и др.) или β-адреноблокаторы. Назначение ингибиторов ангиотензин-превращающего фермента и блокаторов ангиотензиновых рецепторов нежелательно из-за возможности развития гиперкалиемии, снижения скорости клубочковой фильтрации. При эклампсии для получения быстрого гипотензивного эффекта вводят прямые вазодилататоры (гидралазин и др.).
-
Возможно назначение антиагрегантов для улучшения почечного кровотока [дипиридамол, пентоксифиллин (Трентал♠)], антикоагулянтов — при выраженной гиперкоагуляции, связанной с развитием нефротического синдрома.
-
При остром повреждении почек, нарастающей азотемии, неконтролируемой гиперкалиемии необходимо проведение диализа.
Прогноз
Прогноз обычно хороший, большинство (85–90%) детей выздоравливает. У некоторых пациентов возникает быстропрогрессирующий (экстракапиллярный) ГН с развитием прогрессирующей хронической болезни почек.
8.2.2. Быстропрогрессирующий гломерулонефрит
Быстропрогрессирующий ГН (подострый, злокачественный, экстракапиллярный, с полулуниями) характеризуется чрезвычайно высокой активностью, тяжелым прогрессирующим течением, нарастающей почечной недостаточностью с развитием терминальной уремии в течение нескольких недель или месяцев.
Быстропрогрессирующий ГН у детей как вариант первичного ГН наблюдают редко (в 1–2% случаев), преимущественно у подростков; вторичный — может развиться при различных инфекционных, системных заболеваниях, опухолях. Чаще всего это конечная стадия острого постстрептококкового ГН и синдрома Гудпасчера.
Этиология и патогенез
Выделяют несколько иммунопатогенетических типов быстропрогрессирующего ГН в зависимости от наличия или отсутствия иммунных депозитов в клубочках почек, а также характера их свечения при иммунофлюоресцентной микроскопии:
-
с АТ против Аг базальной мембраны клубочков при идиопатическом быстропрогрессирующем ГН, синдроме Гудпасчера (АТ циркулируют в сыворотке крови и выявляются в биоптате почки — линейное свечение);
-
иммунокомплексный, наиболее типичный для острого постстрептококкового ГН, криоглобулинемии, СКВ, IgA-нефропатии, пурпуры Шенляйна–Геноха, мембранопролиферативного ГН; в мезангии и капиллярной стенке обнаруживают депозиты иммунных комплексов, имеющие гранулярное свечение;
-
без иммунных депозитов (малоиммунный), но часто с АТ к компонентам цитоплазмы нейтрофилов (протеиназе 3, миелопероксидазе), которые определяют в сыворотке крови, что характерно для микроскопического полиангиита, гранулематоза Вегенера.
Морфология
Патологоанатомическая особенность этого варианта — пролиферация клеток париетального эпителия (экстракапиллярный) с образованием более чем в 50% клубочков полулуний, служащих гистологическим маркером быстропрогрессирующего ГН (рис. 8.5).
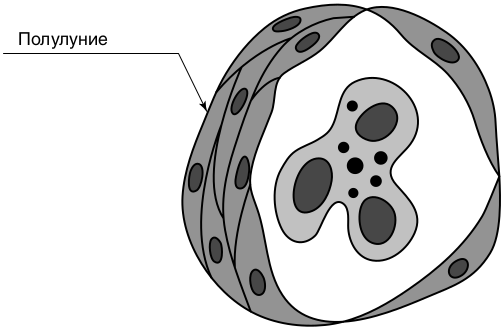
Полулуния — следствие тяжелого повреждения клубочков, вызванного АТ к базальной мембране или иммунными комплексами, или АТ к компонентам цитоплазмы нейтрофилов, приводящего к разрыву стенок капилляров. В результате в пространство капсулы Боумена попадают плазменные белки и воспалительные клетки (в основном пролиферирующие париетальные эпителиальные клетки и макрофаги), фибрин, формирующие инфильтрат с образованием дугообразных утолщений капсулы клубочка — полулуний (получивших такое название из-за характерного внешнего вида на срезах клубочков).
Последние окружают клубочковые капилляры и вызывают их спадение. При преобладании в полулуниях макрофагов, разрыве капсулы из интерстиция поступают фибробласты и миофибриллы, синтезирующие матриксные белки (коллаген типов 1 и 3, фибронектин), что ведет к необратимому фиброзу полулуний.
Клиническая картина и диагностика
Клинические и лабораторные признаки:
-
остронефритический синдром (см. острый постстрептококковый ГН);
-
быстропрогрессирующая почечная недостаточность, критерий которой — повышение уровня креатинина сыворотки в 2 раза за каждые 3 мес болезни или в более короткий срок.
АГ высокая, носит стойкий характер. Протеинурия неселективная, субнефротического уровня, иногда развивается нефротический синдром. Рано, с первых месяцев, а иногда и с первых недель болезни (2–3-й), появляются клинические и лабораторные симптомы почечной недостаточности:
Терминальная уремия развивается в течение года от дебюта заболевания. Результаты иммунологического исследования крови зависят от иммунопатогенетического типа быстропрогрессирующего ГН: при иммунокомплексном отмечают снижение концентрации С3-компонента и гемолитической активности комплемента, при антительном — обнаруживают АТ к базальной мембране (к коллагену 4-го типа), при малоиммунном — АТ к компонентам цитоплазмы нейтрофилов.
Лечение
Лечение быстропрогрессирующего ГН следует начинать безотлагательно ввиду чрезвычайно высокой активности процесса и быстрой необратимой потери функции почек.
Показана интенсивная иммуносупрессивная терапия, которую начинают, не дожидаясь результатов иммунологии крови и нефробиопсии. С этой целью, согласно международному стандарту, используют:
После верификации диагноза лечение глюкокортикоидами сочетают с цитостатиками:
Комбинированная иммуносупрессивная терапия рекомендуется при иммунокомплексном и малоиммунном патогенетических типах быстропрогрессирующего ГН.
При неэффективности лечения и развитии терминальной почечной недостаточности начинают программный гемодиализ и выполняют трансплантацию почки.
Дифференциальную диагностику быстропрогрессирующего ГН необходимо проводить с заболеваниями, приводящими к острому повреждению почек, такими как острый постстрептококковый ГН, острый интерстициальный нефрит, острый тубулярный некроз, атипичный гемолитико-уремический синдром и др.
Прогноз
Прогноз, даже при своевременно начатом активном лечении, остается тяжелым из-за высокого риска быстрого исхода в терминальную почечную недостаточность. Для его оценки необходимо проведение биопсии почек. При быстропрогрессирующем ГН, ассоциированном с острым постстрептококковым ГН, возможен благоприятный исход в ремиссию с восстановлением почечных функций.
8.2.3. Хронический гломерулонефрит
Хронический ГН — группа разнородных первичных гломерулопатий, характеризующихся прогрессирующими, деструктивными, склеротическими изменениями с постепенным ухудшением почечных функций и исходом в хроническую почечную недостаточность.
Пролиферативные гломерулонефриты
Мезангиопролиферативный гломерулонефрит (иммуноглобулин А-нефропатия)
IgA-нефропатия может быть самостоятельным заболеванием (первичная IgA-нефропатия — болезнь Берже) и вторичной — при многих системных (пурпура Шенляйна–Геноха, СКВ и др.) и хронических заболеваниях (ВГВ и гепатит С, ЯК, БК, МВ, саркоидоз и др.).
IgA-нефропатия (болезнь Берже) — самая распространенная форма первичного ГН.
Заболевание может дебютировать в любом возрасте. Мальчики болеют в 2 раза чаще девочек.
Морфология: пролиферация мезангиальных клеток (очаговая или диффузная), расширение мезангия, отложение иммунных комплексов в мезангии и под эндотелием (рис. 8.6). Чаще всего в клубочках выявляют депозиты IgА — IgA-нефропатия, с которой в настоящее время отождествляют данный морфологический вариант ГН.
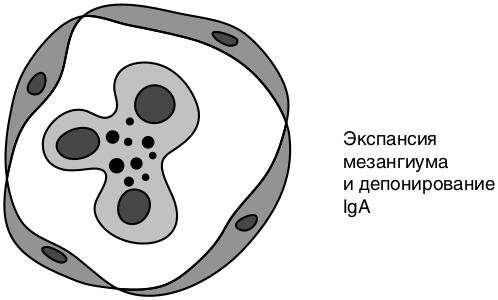
Механизм развития IgA-нефропатии — иммунокомплексный, в развитии которого ведущая роль принадлежит увеличению синтеза и изменению структуры молекулы IgA, вызванному нарушением процессов ее гликозилирования и полимеризации. IgA-полимеры и IgA-содержащие иммунные комплексы длительно находятся в циркуляции, не выводятся клетками ретикулоэндотелиальной системы, депонируются в мезангии, в результате чего активируется синтез клетками почек различных цитокинов, факторов роста, развиваются характерные морфологические изменения.
Клиническая картина IgA-нефропатия разнообразна. Наиболее частые клинические варианты следующие.
-
Макрогематурия — в виде повторных эпизодов (рецидивирующая), сопровождающих респираторную инфекцию (синфарингитная). Макрогематурия возникает одновременно или в первые дни болезни (2–3-й) и сохраняется от нескольких часов до нескольких дней. Моча обычно бурого цвета. Рецидивы могут провоцироваться вакцинацией, физической нагрузкой. Между эпизодами синфарингитной макрогематурии в анализах мочи выявляют микрогематурию.
-
Персистирующая микрогематурия, иногда в сочетании небольшой протеинурией и/или АГ.
При фазово-контрастной микроскопии осадка мочи обнаруживают дисморфные эритроциты, указывающие на гломерулярное происхождение гематурии. У 35–60% больных отмечают повышение уровня IgА в сыворотке крови. Тактика лечения IgA-нефропатия основана на оценке риска прогрессирования заболевания. При изолированной гематурии лечение не назначают. При гематурии, в том числе с эпизодами синфарингитной макрогематурии, небольшой протеинурии (0,5–1 г/сут), нормальной скорости клубочковой фильтрации и отсутствии АГ целесообразна нефропротективная терапия. При протеинурии нефротического уровня показана иммуносупрессивная терапия — глюкокортикоиды в течение 6 мес. Необходима санация очагов инфекции, провоцирующих обострения заболевания.
У детей прогноз IgA-нефропатии (болезни Берже) обычно благоприятный, особенно если она проявляется синфарингитной гематурией.
Дифференциальную диагностику прежде всего следует проводить с наследственными гломерулопатиями — болезнью тонких базальных мембран и синдромом Альпорта. Болезнь тонких базальных мембран (семейная доброкачественная гематурия) — заболевание с аутосомно-доминантным типом наследования, проявляющееся изолированной микрогематурией и имеющее благоприятное течение. Его диагностируют по фокальному или диффузному истончению гломерулярных базальных мембран, которое выявляют при электронной микроскопии нефробиоптатов, иммунный материал в ткани почек отсутствует. Синдром Альпорта (Х-сцепленный, аутосомно-рецессивный и аутосомно-доминантный варианты) — генетически детерминированная гломерулопатия, обусловленная мутацией генов, кодирующих коллаген 4-го типа базальных мембран. Проявляется гематурией и/или протеинурией, имеет прогрессирующее течение с исходом в почечную недостаточность. Поражение почек может сочетаться с двусторонней нейросенсорной тугоухостью и патологией зрения (передний лентиконус, крапинки на желтом пятне и эрозии роговицы).
Мембранопролиферативный гломерулонефрит
Мембранопролиферативный (мезангиокапиллярный) ГН — иммунокомплексное заболевание, морфологические особенности которого — мезангиальная пролиферация и утолщение стенок капилляров с образованием двухконтурных базальных мембран за счет проникновения в них мезангиальных клеток (рис. 8.7). При электронной микроскопии выявляют субэндотелиальные депозиты иммунных комплексов (1-й тип) или плотные отложения иммунных комплексов внутри базальной мембраны клубочков (2-й тип, болезнь плотных депозитов).
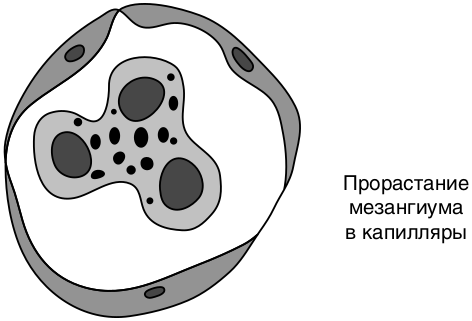
Прогрессирование патологического процесса приводит к развитию склероза и формированию фибропластического ГН — финалу большинства форм хронического ГН. Развивается склероз капиллярных петель клубочка, формируются фиброэпителиальные и фиброзные полулуния, утолщение и склероз капсулы клубочка.
Обычно мембранопролиферативный ГН диагностируют у детей 10–12 лет. Нередко устанавливают связь мембранопролиферативного ГН (1-й тип) с инфицированием вирусом ВГВ и HCV. Этот тип ГН выявляют при СКВ, синдроме Шегрена, саркоидозе, лимфоме.
Особенности морфологии (сочетание пролиферация мезангия с поражением базальных мембран клубочков) определяют клиническую картину мембранопролиферативного ГН — сочетание нефритического и нефротического синдромов. Для первичного и вторичном вариантов заболевания характерны: стойкая, высокая АГ, выраженная неселективная протеинурия, гипокомплементемия со снижением уровня С3- и/или С4-компонентов комплемента, анемия, криоглобулинемия (особенно у больных гепатитом С).
При выборе терапии идиопатического мембранопролиферативного ГН учитывают клиническое течение заболевания и результаты биопсии почки. При ведущем нефротическом синдроме (НС), медленном снижении функции почек рекомендуют циклофосфамид (2–2,5 мг/кг в сут) или микофенолата мофетил в сочетании с преднизолоном в альтернирующем режиме. При нефритическом синдроме с быстропрогрессирующим падением почечных функций вначале проводят плазмаферез, пульс-терапию метилпреднизолоном (3 дня), далее продолжают иммуносупрессивное лечение по предыдущей схеме.
Мембранопролиферативный ГН — один из самых неблагоприятных вариантов ГН с быстрым исходом (у 50% больных) в терминальную почечную недостаточность.
Непролиферативные гломерулонефриты
Непролиферативные ГН обусловлены иммунопатологическими процессами, но при этом нет морфологических признаков воспаления, поскольку пролиферации собственных клеток клубочка отсутствует или выражена минимально. Поэтому в ряде классификаций их относят к группе непролиферативных гломерулопатий.
Подоцитопатии — болезнь минимальных изменений и фокально-сегментарный гломерулосклероз (особенно в дебюте заболевания) — имеют сходную морфологическую картину, что позволило объединить их термином идиопатический нефротический синдром.
Нефротический синдром — симптомокомплекс, включающий высокую протеинурию (более 50 мг/кг в сут), гипоальбуминемию (ниже 25 г/л), гиперлипидемию и отеки. Распространенность его составляет 1 случай на 6000 детей, у мальчиков в 2 раза чаще, чем у девочек.
Болезнь минимальных изменений
Болезнь минимальных изменений имеет ряд синонимов: идиопатический нефротический синдром, ГН с минимальными изменениями, НС с минимальными изменениями, стероид-чувствительный НС, исторический термин — липоидный нефроз.
Болезнь минимальных изменений наблюдают в 80–90% случаев НС у детей младшей возрастной группы (до 10 лет), в 50% — у подростков. В структуре всех морфологических вариантов ГН у детей составляет 76,6%.
Морфология. Изменения клубочков выявляют только на ультраструктурном уровне (электронная микроскопия) в виде диффузного слияния (сглаживания) ножковых отростков подоцитов (подоцитопатия) и их микровиллезной трансформации (появления на эпителиальной поверхности многочисленных ворсинчатых образований, направленных в мочевое пространство); при световой микроскопии клубочек выглядит неизмененным, при флюоресцентном исследовании отсутствуют отложения Ig и фракций комплемента в структурах нефрона (рис. 8.8).
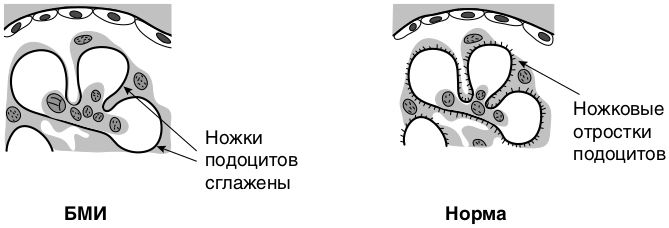
Болезнь минимальных изменений чаще всего бывает идиопатической.
Возможны вторичные формы, развивающиеся на фоне установленных причин: инфекций, лекарственных (чаще всего НПВП), токсических воздействий, атопии, злокачественных заболевания и др.).
Патогенетические механизмы идиопатической болезни минимальных изменений:
-
дисфункция Т-клеточного звена иммунитета, приводящая к продукции «фактора проницаемости», вызывающего дезорганизацию цитоскелета подоцитов, следствием чего становится увеличение диффузии альбумина через гломерулярную базальную мембрану (ГБМ); механизмы активации клеточного иммунитета и сам «фактор проницаемости» остаются неизвестными;
-
генетически детерминированные изменения протеинов щелевой диафрагмы и актинового цитоскелета подоцитов, вследствие чего повышена проницаемость гломерулярного фильтра.
Клинические и лабораторные симптомы идиопатической болезни минимальных изменений следующие.
-
Внезапное появление НС без видимой причины. Иногда провоцирующими факторами могут быть атопические реакции, ОРИ.
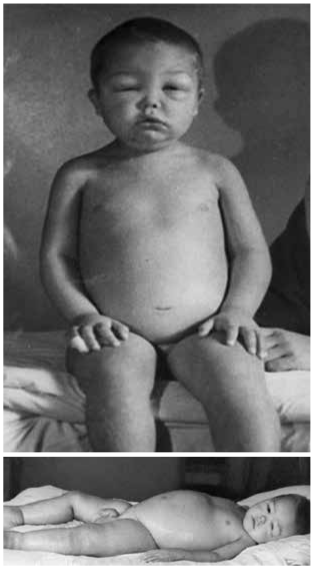
-
Генерализованные отеки (рис. 8.9), часто прогрессирующие до анасарки с развитием асцита, гидроторакса, гидроперикарда. Кожа бледная, холодная на ощупь. Диурез снижен.
-
Протеинурия массивная (может достигать 20–30 г в сут и более), высокоселективная (альбуминурия).
-
Резкая гипоальбуминемия (не всегда пропорциональная выраженности протеинурии), определяющая тяжесть НС, и гиповолемия.
Потеря белка (альбумина) с мочой приводит к снижению онкотического давления плазмы крови. Возникает онкотический градиент между внутрисосудистой и экстравазальной средой, вследствие чего происходит перемещение жидкости из сосудистого русла в ткани. Для восстановления объема циркулирующей крови запускаются компенсаторные механизмы (секретируется антидиуретический гормон, ингибируется синтез предсердного натрийуретического фактора, активируются ренин-ангиотензин-альдостероновая система (РААС), симпатическая нервная система), стимулирующие задержку натрия и воды в почках. Гипоальбуминемия усиливается (гипоальбуминемия разведения) и поддерживает отеки.
-
Выраженная гиперлипидемия с дислипопротеинемией (повышение концентрации общего холестерина, триглицеридов, липопротеидов низкой плотности, липопротеина А и снижение концентрации липопротеинов высокой плотности), причинами которых считают повышенный синтез липопротеидов низкой плотности, вызванный снижением онкотического давления, потерю с мочой липорегуляторных субстанций и снижение катаболизма липидов.
-
Симптомокомплекс НС не сопровождается ни АГ, ни гематурией («чистый» НС). Очень редко отмечают кратковременное повышение АД, связанное с задержкой натрия в ответ на гиповолемию, и микрогематурию.
Возможно развитие угрожающих жизни осложнений.
-
Гиповолемический шок, связанный с резким значительным падением объема циркулирующей крови: боли в животе, рвота, кожная эритема, гипотония, тахикардия, холодные конечности, уменьшение количества мочи.
-
Острое повреждение почек (см. острый постстрептококковый ГН), главная причина которого — гипоперфузия почек из-за гиповолемии.
-
Тромбозы венозные (почечных вен, глубоких вен нижних конечностей и др.), наблюдаемые чаще, и артериальные. Предполагают, что развитие их связано с гемоконцентрацией, повышением вязкости крови на фоне гиповолемии, гиперфибриногенемией, повышением агрегации тромбоцитов, потерей с мочой плазминогена, антитромбина III, протеинов С и S, угнетением фибринолиза. Способствуют их возникновению обездвиженность, лечение глюкокортикоидами и диуретиками.
-
Тяжелые инфекции вследствие вторичного иммунодефицита, вызванного потерей Ig и компонентов системы комплемента с мочой, дисфункцией Т-клеточного звена иммунитета, общих метаболических нарушений, применения иммуносупрессорных препаратов.
Наряду с этим развивается белково-энергетическая недостаточность, главная причина которой — гипоальбуминемия.
Течение болезни может быть острым с исходом в стойкую ремиссию и рецидивирующим.
Лечение. В активном периоде заболевания показан постельный режим. Объем жидкости дозируют в зависимости от диуреза. Назначают гипохлоридную диету или бессолевой стол. При резкой гиперлипидемии необходимо сократить прием жиров.
Диуретические препараты: петлевые диуретики (фуросемид, торасемид и др.).
Стандартное лечение первого эпизода болезни минимальных изменений [клинические рекомендации KDIGO (Kidney Disease: Improving Global Outcomes, научного общества нефрологов России)] — глюкокортикоидная терапия по схеме:
-
преднизолон в дозе 60 мг/м2 поверхности тела или 2 мг/кг (максимально 60 мг/сут) внутрь ежедневно в течение 4–6 нед; к этому времени у большинства детей наступает полная или частичная ремиссия (стероид-чувствительный НС);
-
снижение суточной дозы до 40 мг/м2 с переходом на альтернирующий режим (прием препарата через день), который используют в течение 4–6 нед;
-
продолжение постепенного снижения суточной дозы препарата до полной отмены.
Общая длительность терапии глюкокортикоидами должна составлять 4–5 мес.
Стероид-чувствительность при болезни минимальных изменений у детей составляет более 90%.
Поэтому для назначения инициальной терапии болезни минимальных изменений нет необходимости в предварительном проведении биопсии почки. Она возникает при подозрении на иные морфологические изменения в почечной ткани или вторичный характер болезни минимальных изменений.
Рецидив, развившийся после отмены глюкокортикоидов, свидетельствует о стероид-зависимости. Лечение первого рецидива НС проводят так же, как терапию в дебюте болезни. Отсутствие эффекта от терапии глюкокортикоидами в течение 8 нед указывает на резистентность НС к глюкокортикоидам. При частых рецидивах НС, стероид-зависимости и стероид-резистентности применяют стероидсберегающие препараты — алкилирующие агенты (циклофосфамид или хлорамбуцил), сохраняя прием самых низких доз преднизолона, необходимых для поддержания ремиссии.
При отсутствии терапевтического эффекта используют левамизол, ингибиторы кальцинейрина (циклоспорин или такролимус), микофенолата мофетил, ритуксимаб. При стероид-резистентном НС возможно проведение пульс-терапии метилпреднизолоном или комбинированной терапии различными иммуносупрессантами по индивидуальным схемам. Показано длительное применение ренопротективных препаратов [ингибиторов ангиотензин-превращающего фермента (фозиноприла, эналаприла)] и блокаторов ангиотензиновых рецепторов.
При угрозе тромботических осложнений (выраженный НС с гиповолемией, гиперфибриногенемией и гиперлипидемией) проводят профилактическое лечение антикоагулянтами: гепарины [далтепарин натрия (Фрагмин♠), надропарин кальция (Фраксипарин♠)], варфарин.
Показания к биопсии почки у детей с НС:
-
возраст ребенка до 1 года, когда велика вероятность генетических вариантов НС;
-
стероид-резистентность, которую констатируют при отсутствии эффекта от терапии глюкокортикоидами в дебютной дозе в течение 8 нед;
-
стероид-зависимость, свидетельство которой — рецидив НС при снижении дозы преднизолона или в течение 2 нед после его отмены;
Прогноз при наличии чувствительности к глюкокортикоидам благоприятный — у большинства детей развивается стойкая ремиссия. Неблагоприятными факторами следует считать генетически обусловленную болезнь минимальных изменений, а также первичную и вторичную (отсутствие эффекта от глюкокортикоидов при рецидивах НС) стероид-резистентность, которые могут быть связаны с наличием фокально-сегментарного гломерулосклероза или трансформацией минимальных изменений в данную патологию.
Фокально-сегментарный гломерулосклероз
Фокально-сегментарный гломерулосклероз — главная причина (более 50% случаев) резистентности к глюкокортикоидам у детей.
Морфология: при световой микроскопии в начале заболевания изменения трактуют как минимальные (подоцитопатия), в последующем выявляют зоны склероза и гиалиноза в некоторых сегментах (сегментарные) отдельных (фокальные) клубочков (рис. 8.10).
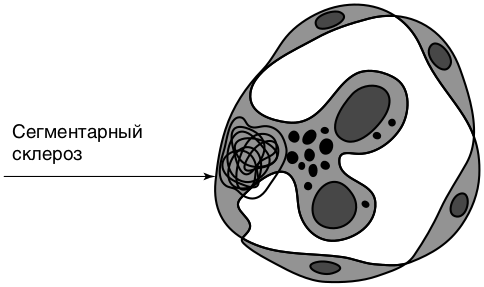
Сходство морфологии фокально-сегментарного гломерулосклероза и болезни минимальных изменений позволило предположить, что это разные стадии или разной тяжести варианты одного заболевания.
В развитии первичного варианта фокально-сегментарного гломерулосклероза ведущая роль принадлежит генетическому дефекту или действию циркулирующих факторов проницаемости (кардиотропиноподобному цитокину 1 из семейства ИЛ-6, растворимому рецептору к урокиназе и др.). Подоциты, подвергшиеся трансдифференциации, приобретают свойства макрофагов и участвуют в формировании очагов фиброза (скероза).
Клинико-лабораторные особенности фокально-сегментарного гломерулосклероза — НС или персистирующая субнефротическая протеинурия в сочетании с микрогематурией и АГ.
Основу терапии идиопатического фокально-сегментарного гломерулосклероза с НС составляют глюкокортикоиды (см. лечение болезни минимальных изменений). Эффективность терапии повышает применение высокодозных внутривенных пульсов метилпреднизолона.
Фокально-сегментарный гломерулосклероз — основная форма гломерулопатии, при которой развивается терминальная почечная недостаточность, требующая заместительной почечной терапии (программного диализа).
Прогноз неблагоприятный, особенно у детей с массивной протеинурией (более 10 г/сут). Кроме того, фокально-сегментарный гломерулосклероз очень часто (30–50%) рецидивирует в трансплантированной почке.
Мембранозная нефропатия
Мембранозная нефропатия в структуре НС у детей составляет менее 1%. Чаще развивается у детей школьного возраста (6–15 лет). Для мембранозной нефропатии характерно диффузное утолщение гломерулярной базальной мембраны (рис. 8.11) и изменение ее структуры из-за отложения иммунных депозитов под подоцитами (субэпителиально) и внутри мембраны (интрамембранозно).
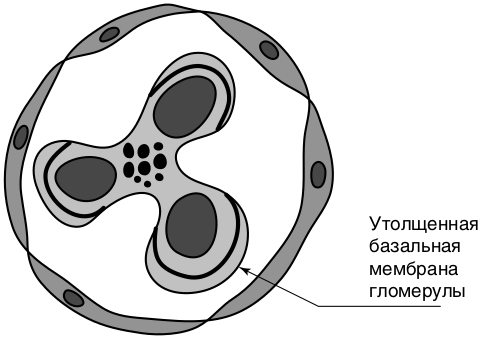
Механизм развития мембранозной нефропатии иммунокомплексный. Основа формирования иммунных комплексов при первичном форме патологии — образование аутоантител к внутренним Аг клубочка — подоцитарному трансмембранному рецептору фосфолипазы. Основные клинические проявления — НС или субнефротическая протеинурия.
При мембранозной нефропатии самый высокий риск тромботических осложнений НС. Возможны микрогематурия, АГ.
У детей заболевание чаще протекает без НС с нормальной функцией почек, часто приводит к развитию полной спонтанной ремиссии и не требует иммуносупрессивной терапии. Предикторы неблагоприятного прогноза — НС с массивной протеинурией, АГ и снижение почечных функций.
Тестовые задания
Ответы: 1 — b; 2 — a; 3 — a; 4 — d.
Литература
-
Детская нефрология : практическое руководство / под ред. Э. Лойманна, А.Н. Цыгина, А.А. Саркисяна. М. : Литтерра, 2010.
-
Нефрология. Клинические рекомендации / под ред. Е.М. Шилова, А.В. Смирнова, Н.Л. Козловской. М. : ГЭОТАР-Медиа, 2016.
-
О’Каллагхан К.А. Наглядная нефрология. М. : ГЭОТАР-Медиа, 2009.
-
Эрман М.В. Нефрология детского возраста : руководство для врачей. СПб. : СпецЛит, 2010.