РОССИЙСКОЕ РЕСПИРАТОРНОЕ ОБЩЕСТВО
РЕСПИРАТОРНАЯ МЕДИЦИНА
Респираторная медицина: руководство: в 3 т. / под ред. А. Г. Чучалина. — 2-е изд., перераб. и доп. — М.: Литтерра, 2017. — Т. 3. — 464 с. |
Руководство в трех томах
Под редакцией академика РАН А.Г. Чучалина
2-е издание, переработанное и дополненное
Том 3
Посвящается памяти Сергея Петровича Боткина
Москва
Издательство «Литтерра»
2017
УДК 616.2
ББК 54.12
Р43
04-УПС-0294
Р43
Респираторная медицина: руководство: в 3 т. / под ред. А. Г. Чучалина. — 2-е изд., перераб. и доп. — М.: Литтерра, 2017. — Т. 3. — 464 с.: ил.
ISBN 978-5-4235-0272-0 (т. 2)
ISBN 978-5-4235-0273-7 (общ.)
В третьем томе руководства представлены клинические разделы, посвященные интерстициальным и редким заболеваниям легких, внешним воздействиям и легочным проявлениям болезней других органов и систем, регуляции дыхания, дыхательной недостаточности и реабилитации пациентов.
Издание рассчитано на широкий круг специалистов: терапевтов, пульмонологов, фтизиатров, аллергологов, онкологов и других специалистов разной степени квалификации и подготовки. Третий том представляет интерес также для реаниматологов, ревматологов, профпатологов, врачей-реабилитологов.
Изучение руководства позволит врачам достичь высокого уровня компетенции и будет способствовать решению сложнейших вопросов клинической практики.
УДК 616.2
ББК 54.12
Права на данное издание принадлежат ООО «Издательство «Литтерра». Воспроизведение и распространение в каком бы то ни было виде части или целого издания не могут быть осуществлены без письменного разрешения ООО «Издательство «Литтерра».
ISBN 978-5-4235-0272-0 (т. 3)
ISBN 978-5-4235-0273-7 (общ.)
15.7. Альвеолярный протеиноз
М.М. Илькович
Альвеолярный протеиноз (синонимы: альвеолярный липопротеиноз, альвеолярный фосфолипидоз, легочный альвеолярный фосфолипопротеиноз) — редкое заболевание, характеризующееся накоплением в альвеолах белков и липидов сурфактанта, нарушением газообмена и прогрессированием ДН. Болезнь впервые описана S.H. Rosen и соавт. в 1958 г. [2].
Эпидемиология
АП относится к редким (орфанным) заболеваниям и встречается с частотой от 1 до 4 случаев на 1 млн взрослых, преимущественно — у лиц среднего возраста (20–50 лет), причем у мужчин чаще, чем у женщин (3:1) [3]. Описаны случаи заболевания у детей и лиц пожилого возраста.
Этиология, факторы риска
Выделяют первичную, вторичную или врожденную формы АП. В 90% случаев заболевание является первичным, или идиопатическим. Этиология его неизвестна. Вторичный АП развивается вследствие ряда причин: гематологические злокачественные заболевания, иммунодефицитные состояния, хронические инфекции. При этих состояниях нарушается функция альвеолярных макрофагов, что ведет к избыточному накоплению сурфактанта в альвеолах [4]. Врожденная форма обусловлена мутациями генов, кодирующих структуру белков сурфактанта В и С, гена АВСА1-транспортера, а также рецептора гранулоцитарно-моноцитарного колониестимулирующего фактора (ГМ-КСФ) [4].
Курение, по-видимому, является фактором риска развития АП. Так, курильщики, в том числе бывшие, составляют от 56 до 79% всех пациентов с этим заболеванием [1, 5]. Можно предположить, что большая распространенность АП среди лиц мужского пола в значительной степени может быть объяснена большей распространенностью курения среди мужчин [6]. Воздействие таких веществ, как углеводороды, кадмий, титан, асбест, алюминий, имеет место приблизительно у половины больных АП.
Патогенез
Прогресс в понимании патогенеза АП произошел в 1994 г., когда было доказано, что у мышей, нокаутных по гену ГМ-КСФ или его рецептору (у таких животных в результате прицельного разрушения определенного гена не синтезируется соответствующий белковый продукт), нарушен гомеостаз сурфактанта и в легких выявляются изменения, сходные с проявлениями АП [7]. Более того, было доказано, что применение экзогенного ГМ-КСФ у этих животных приводит к нормализации катаболизма сурфактанта и значительно снижает выраженность патологических изменений в легких. Таким образом, было подтверждено, что именно ГМ-КСФ обеспечивает процесс клиренса (реутилизации) сурфактанта.
Сурфактант играет важнейшую роль в снижении поверхностного натяжения в альвеолах, предотвращая спадение альвеол и транссудацию капиллярной жидкости в альвеолярное пространство. Около 90% сурфактанта составляют липиды (преимущественно — фосфолипиды), 10% — белки, менее 1% — углеводы. Четыре вида белков сурфактанта — SP-A, SP-B, SP-C и SP-D — вносят вклад в его структурные и поверхностно активные свойства, участвуют в опсонизации микробных патогенов и стимулируют защитные функции альвеолярных макрофагов. Липиды и белки сурфактанта синтезируются, накапливаются и секретируются в альвеолы альвеолоцитами II типа; утилизируются ими же, а также альвеолярными макрофагами. Ключевую роль в этом процессе играет ГМ-КСФ — полипептидный цитокин с молекулярным весом 23 кД, продуцируемый В-лимфоцитами. Он является основным фактором роста и дифференцировки гемопоэтических клеток (гранулоцитов, макрофагов, эозинофилов), взаимодействуя с рецепторами на их поверхности. Рецепторы для ГМ-КСФ имеются также на клеочной стенке альвеолоцитов II типа. Действуя опосредованно через транскрипционный фактор PU.1, ГМ-КСФ обеспечивает процесс катаболизма сурфактанта альвеолярными макрофагами.
У новорожденных с признаками врожденного АП обычно выявляются определенные генетические причины избыточного накопления сурфактанта в легких: чаще всего это мутации генов, кодирующих структуру белков сурфактанта (SP-В, SP-С), либо ГМ-КСФ или его рецептора (ГМ-КСФ/ ИЛ-3/ИЛ-5) [6]. Однако у взрослых больных АП убедительных доказательств связи развития заболевания с подобными мутациями не получено: структура гена ГМ-КСФ, его рецептора ГМ-КСФ и уровень мРНК ГМ-КСФ не изменены, ответ альвеолярных макрофагов на действие ГМ-КСФ также не нарушен. В 1999 г. японские исследователи Nakata и соавт. [8] обнаружили в сыворотке крови, а также в жидкости БАЛ больных АП антитела (IgG) против ГМ-КСФ, отсутствующие как у доноров, так и у больных другими заболеваниями легких. Это в большинстве случаев объясняет механизм нарушения ГМ-КСФ-зависимого клиренса сурфактанта при АП. Таким образом, есть основания полагать, что идиопатический АП является аутоиммунным заболеванием и связан с подавлением функции ГМ-КСФ аутоантителами, вследствие чего нарушается клиренс сурфактанта и происходит избыточное его накопление в альвеолах. Накопление сурфактанта, в свою очередь, ингибирует функцию альвеолярных макрофагов, что в дальнейшем еще более подавляет его клиренс.
Патологическая анатомия
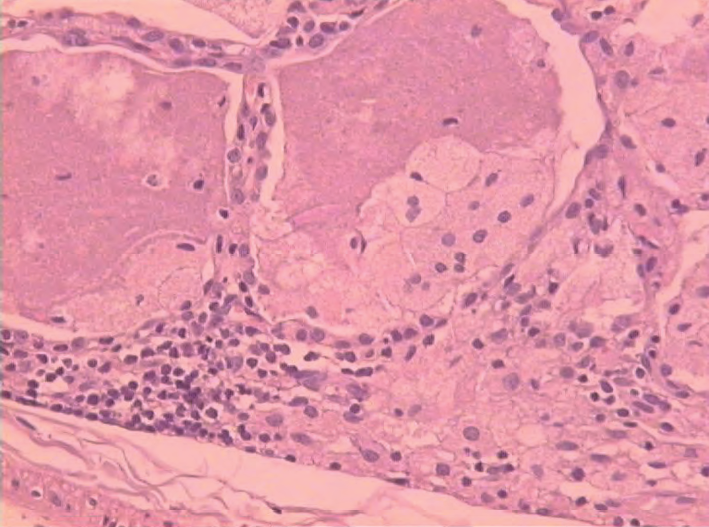
На поверхности легких определяются серовато-белые плотные бугорки в виде зерен. Альвеолярные пространства и респираторные бронхиолы заполнены гранулярным ацидофильным содержимым. Альвеолярные перегородки обычно не изменены. Признаков воспаления или фиброза не обнаруживается, однако отмечается гиперплазия альвеолоцитов II типа. Материал, содержащийся в альвеолах, включает сурфактантоподобное вещество. Это фосфолипиды, которые дают яркий пурпурный цвет при окрашивании реактивом Шиффа (PAS-реакция) и не окрашиваются Alcian blue (рис. 15.57) [1]. Близлежащие участки легочной ткани поражаются неравномерно: часть альвеол может быть заполнена белковолипидным материалом, а в соседних участках могут обнаруживаться лишь мелкие вкрапления белкового вещества; встречаются и абсолютно интактные (непораженные) участки.
Клиническая картина
В течение длительного времени заболевание протекает бессимптомно и нередко выявляется случайно при профилактическом флюорографическом исследовании. Ведущим клиническим признаком болезни является медленно прогрессирующая одышка, которая может сопровождаться сухим или со скудной мокротой кашлем, субфебрильной температурой, болями в груди, похуданием, быстрой утомляемостью, изредка — кровохарканьем. При прогрессировании ДН отмечается цианоз. Периоды обострения болезни, сопровождающиеся ухудшением общего самочувствия и лихорадкой, следует рассматривать, вероятно, как присоединение суперинфекции, а не как обострение основного заболевания [1].
Течение болезни, как правило, хроническое, однако описаны и острые формы [10]. По мере прогрессирования хронической формы АП усиливается цианоз, формируются «пальцы Гиппократа». Похудание — непостоянный симптом. Из осложнений следует отметить присоединение бактериальной или грибковой суперинфекции, развитие ЛГ и формирование в терминальной стадии легочного сердца. Туберкулез осложняет течение АП в 3–5% случаев.
Диагностика
Патогномоничные клинические признаки при АП отсутствуют, вследствие чего срок между началом заболевания и установлением диагноза нередко составляет несколько лет [1].
При физикальном обследовании больных определяется укорочение перкуторного тона преимущественно над нижними легочными полями. Аускультация выявляет ослабленное везикулярное дыхание, иногда — нежные крепитирующие хрипы.
Лабораторные исследования
Данные общих лабораторных исследований (клинический анализ крови, показатели иммунного и биохимического статуса) неспецифичны. Содержание в сыворотке крови SP-A, SP-B, SP-D, C-реактивного белка, лактатдегидрогеназы, фактора Krebs von den Lungen 6, муциноподобного протеина может повышаться и коррелировать со степенью тяжести заболевания, однако эти признаки неспецифичны и встречаются и при других заболеваниях легких [3]. Степень гипоксемии также зависит от тяжести заболевания. Однако жидкость БАЛ микроскопически и даже внешне имеет характерные особенности при АП. Это маслянистая, непрозрачная, молочно-белая, иногда желтоватая жидкость, которая образует белый осадок при отстаивании. В жидкости БАЛ обнаруживается значительное количество PAS-положительных, эозинофильных бесклеточных телец и альвеолярных макрофагов, содержащих гранулярный эозинофильный материал в фаголизосомах или цитоплазме. У пациентов с идиопатическим АП в сыворотке крови и жидкости БАЛ выявляются антитела к ГМ-КСФ. Обнаружение их с помощью иммуноферментного анализа ELISA является патогномоничным для АП [11]. Кроме того, повышено содержание белков сурфактанта SP-A, SP-D, что выявляется при помощи иммуногистохимических методов. При электронной микроскопии выявляются альвеолярные макрофаги, заполненные фаголизосомами, комплексными включениями, ламеллярными тельцами, каплями холестерола липидов [9]. Концентрические ламеллярные тельца, содержащие фосфолипиды, тубулярный миелин и миелиновые структуры в альвеолярных пространствах и в жидкости БАЛ, являются патогномоничными для АП. Обнаружение антител к ГМ-КСФ в сыворотке крови и/или жидкости БАЛ в большинстве случаев позволяет установить правильный диагноз, не прибегая к более инвазивным методам диагностики. Чувствительность и специфичность этого метода приближаются к 100% [11].
Лучевая диагностика
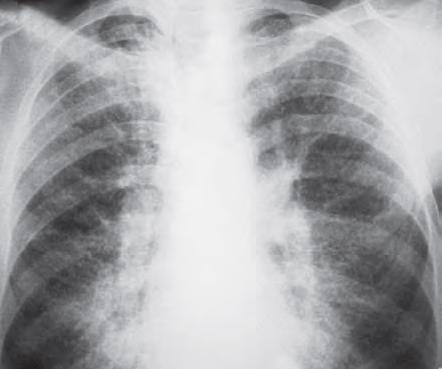
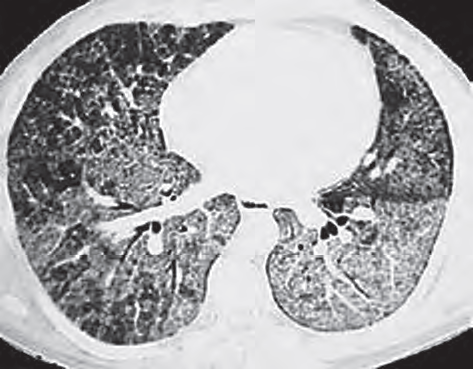
На рентгенограммах у больных АП определяются мелкоочаговые затемнения, имеющие тенденцию к слиянию (рис. 15.58). Изменения чаще двусторонние, симметричные, преимущественная их локализация — средние и нижние легочные поля. Асимметричные или односторонние изменения встречаются в 15–20%. КТВР помогает детализировать выявленные изменения. Утолщенные междольковые перегородки при АП имеют полигональную форму, что дало симптому название «булыжная мостовая», или crazy paving (рис. 15.59). Участки «матового стекла» обычно отграничены от неизмененных областей, поэтому изменения называют «географическими». Симптомы «географической карты» и «булыжной мостовой» очень характерны для АП. Нередко корреляция между клиническими и рентгенологическими данными отсутствует: выраженные рентгенологические изменения могут сопровождаться лишь скудной клинической симптоматикой. В далеко зашедших стадиях заболевания могут выявляться распространенные фиброзные изменения [1].
Показатели функции внешнего дыхания
Показатели ФВД могут оставаться в пределах нормы в течение длительного времени, но по мере прогрессирования болезни выявляется тенденция к формированию рестриктивного синдрома: снижается ДСЛ, усиливается гипоксемия.
Фибробронхоскопия
Фибробронхоскопия не выявляет никаких признаков, характерных для АП, однако исследование лаважной жидкости часто помогает поставить правильный диагноз. В жидкости БАЛ патогномоничны многократное (в 10–100 раз) увеличение содержания белка, выявление бесклеточной глобулярной субстанции, имеющей положительную PAS-реакцию, а также наличие антител против ГМ-КСФ. При исследовании биоптата легочной ткани в альвеолах также выявляется материал, дающий пурпурный или лилово-красный цвет при окраске его реактивом Шиффа. При электронной микроскопии патогномоничным для АП является обнаружение в альвеолах и альвеолярных макрофагах сурфактанта в виде пластинчатых (ламеллярных) телец, однако этот метод исследования в настоящее время редко используется для подтверждения диагноза АП.
Дифференциальная диагностика
Дифференциальную диагностику следует проводить в первую очередь со вторичным протеинозом, являющимся осложнением других заболеваний. Помимо наличия основного (гематологического, онкологического) заболевания, вторичный АП отличается от первичного гранулярным (очаговым) окрашиванием содержащегося в альвеолах PAS-положительного вещества, в то время как для первичного протеиноза характерно равномерное окрашивание.
АП следует дифференцировать с саркоидозом, диссеминированным туберкулезом легких, нередко проявляющимся субклинически, а также с ПЦП. Изучение рентгенологического архива и выявление лимфаденопатии средостения на начальных этапах болезни позволяет исключить АП, для которого увеличение лимфатических узлов средостения не характерно. Проведение целого ряда исследований (микробиологических, серологических, эндоскопических, цитологических, лучевых) в большинстве случаев позволяет исключить или подтвердить специфическую природу заболевания. Трудности диагностики нередко являются причиной ошибочной лечебной тактики. Нередки случаи, когда выявляемые при профилактическом флюорографическом исследовании мелкоочаговые (мелкоточечные) затемнения (иногда сливающиеся и создающие картину инфильтрации) с обеих сторон расцениваются врачом как двусторонняя пневмония. И это несмотря на отсутствие каких-либо жалоб, хотя бы отдаленно напоминающих пневмонию, нормальные показатели клинического анализа крови, нормальную температуру тела. Нередко после длительного безуспешного применения антибиотиков диагноз пересматривается в пользу специфического процесса, что влечет за собой назначение длительной противотуберкулезной химиотерапии. В ряду ошибочных диагнозов можно отметить также идиопатический фиброзирующий альвеолит, причем этот диагноз зачастую ставится лицам со случайно выявленными изменениями в легких, не жалующимся на одышку, и влечет за собой необоснованное назначение системных кортикостероидов. Несомненно, длительное ошибочное лечение негативно отражается на состоянии пациентов: часто отмечаются нежелательные явления от необоснованной лекарственной терапии (гепатотоксичнось, синдром Кушинга и др.), усиливается выраженность изменений в легких при контрольных лучевых исследованиях, прогрессирует ДН.
Формулировка диагноза
АП, хроническое (острое) течение. ДН (степень). ЛГ (степень). Легочное сердце (течение, компенсация).
Лечение
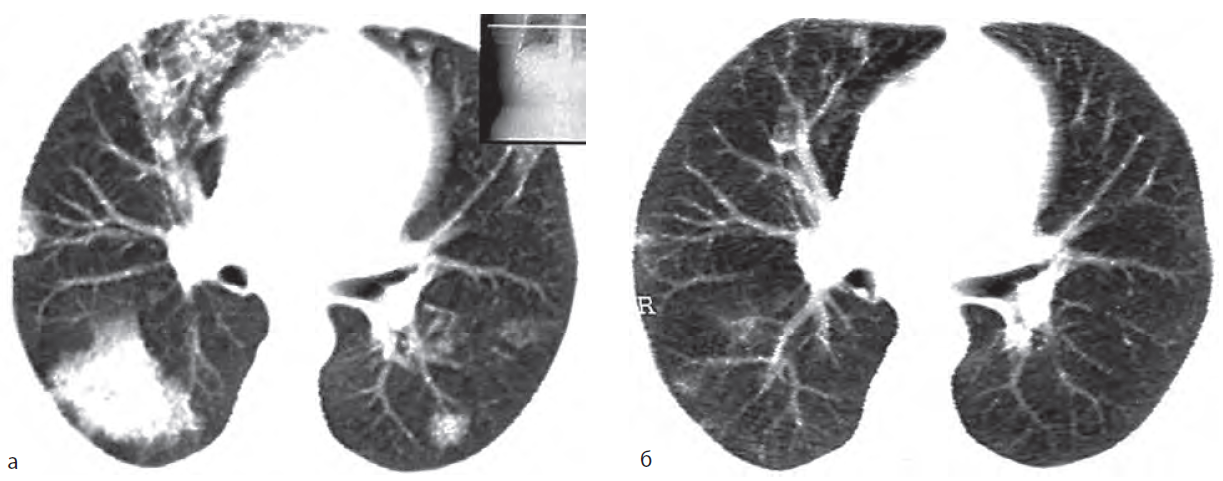
До недавнего времени единственным эффективным методом лечения больных протеинозом был лечебный тотальный БАЛ, применяемый с 1965 г. В настоящее время этот метод лечения также широко распространен и является общепринятым, однако разрабатываются и апробируются новые варианты медикаментозной терапии (см. далее). Процедура тотального БАЛ проводится под общим обезболиванием. Одно легкое вентилируется кислородной смесью через двухпросветную трубку, а второе легкое (доля, сегменты) промывается теплым (температуры тела) стерильным изотоническим раствором натрия хлорида. Возможно добавление к раствору ацетилцистеина. Общий объем жидкости зависит от объема промываемого участка (сегмент, доля, легкое) и составляет от 1 до 10 л и более. В результате эффективно проведенного тотального БАЛ получают мутную жидкость с образованием после отстаивания осадка беловатого цвета (рис. 15.60). Лечебный тотальный БАЛ — процедура высокоэффективная. Клиническое, функциональное и рентгенологическое улучшение отмечается у 75–95% больных [1, 9] (рис. 15.61). Как показывает опыт, процедура тотального БАЛ в достаточной мере безопасна. Возможные осложнения: пневмоторакс, отек легких, бронхоспазм, утяжеление ДН, аспирационная пневмония. Дать конкретные рекомендации о частоте тотального БАЛ трудно, так как у некоторых пациентов после первой процедуры отмечается длительное улучшение и даже выздоровление, у других больных белково-липоидное вещество накапливается вновь, однако с разной скоростью. Длительная ремиссия после однократного лечебного тотального БАЛ, по данным литературы, отмечается в 20–50% случаев.

В случаях выраженной ДН, противопоказаний к проведению вмешательства под общим наркозом, вместо процедуры лечебного тотального БАЛ может выполняться процедура санационной бронхоскопии с проведением сегментарного БАЛ. При ее проведении также применяется теплый изотонический раствор натрия хлорида с добавлением ацетилцистеина. Эффективность процедуры достаточно высока по данным контрольных лучевых исследований. Аналогичная процедура сегментарного БАЛ выполняется и в случаях, когда изменения в легких локальны. Процедура сегментарного лаважа при необходимости может повторяться 1 раз в 2–4 дня на протяжении 7–10 дней.
В последнее десятилетие активно разрабатываются новые, менее инвазивные подходы к терапии этого заболевания. В частности в лечении аутоиммунного (идиопатического) АП проходит клинические исследования рекомбинантный ГМ-КСФ. Предварительные результаты его подкожного и ингаляционного применения можно считать обнадеживающими: препарат применялся у 94 больных АП с положительным эффектом в 58,6% случаев. Однако отмена препарата с большой вероятностью (29,7%) приводит к рецидиву заболевания [12].
Имеется ограниченный опыт применения ритуксимаба — препарата, содержащего синтетические моноклональные антитела к CD20 антигену В-лимфоцитов. Его применение в дозе 1000 мг 1 раз в 14 дней у 10 больных АП привело к улучшению клинико-функциональных показателей в 7 из 9 случаев при удовлетворительной переносимости терапии [13]. Кроме того, поскольку основным патогенетическим механизмом развития заболевания считают выработку аутоантител против ГМ-КСФ, надежды возлагают также на экстракорпоральные методы лечения. Проведение до 10 сеансов мембранного плазмафереза может улучшить состояние пациента, снизить титр аутоантител против ГМ-КСФ, увеличить промежуток времени до проведения следующего тотального БАЛ [14]. Кроме того, имеются отдельные сообщения об успешном применении амброксола при АП [15]. Этот препарат применялся у пациентов старческого возраста, когда другие методы лечения были противопоказаны из-за возможных побочных эффектов. Амброксол применялся в суточной дозе 45 мг на протяжении длительного периода времени (более года) и приводил к улучшению состояния и положительной динамике изменений по данным лучевых исследований.
Лечение врожденных форм АП поддерживающее, хотя имеются отдельные сообщения об успешной трансплантации легких [3]. Терапия вторичного АП подразумевает лечение основного заболевания. В этих случаях целесообразность выполнения лечебного тотального БАЛ представляется сомнительной.
Прогноз
Прогноз при АП, как правило, благоприятный: течение болезни доброкачественное, очень медленно прогрессирующее. Спонтанная ремиссия наблюдается в 10–30% случаев. Шансы на спонтанную ремиссию повышаются у пациентов с недавно возникшими изменениями в легких (до двух лет), при условии быстрого установления правильного диагноза и отказе от курения. Анализ отдаленных результатов лечения 343 больных АП показал, что 5-летняя выживаемость составила около 75% [3]; по нашим данным, она составила 100% [1]. Летальные исходы обусловлены, прежде всего, прогрессированием ДН. В 20% случаев пациенты погибают вследствие бактериальной или грибковой суперинфекции. Прогноз ухудшают поздняя диагностика, а также длительное безуспешное лечение больных антибактериальными препаратами, кортикостероидами, физиотерапевтическими процедурами.
Список литературы
См. 